
Convenient Genetic Encoding of Phenylalanine Derivatives through Their α-Keto Acid Precursors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of Keto-Acids
2.3. Bacterial Strains, Plasmids
2.4. Construction of E. coli C321.ΔA.expΔPBAD Defective in Arabinose Metabolism
2.5. Fluoresce Tests for eGFP Containing ncAAs
2.6. Optimization of GFP Production
2.7. eGFP Purification for MS Detection
3. Results
3.1. Construction of Escherichia coli Strain C321.ΔA.expΔPBAD
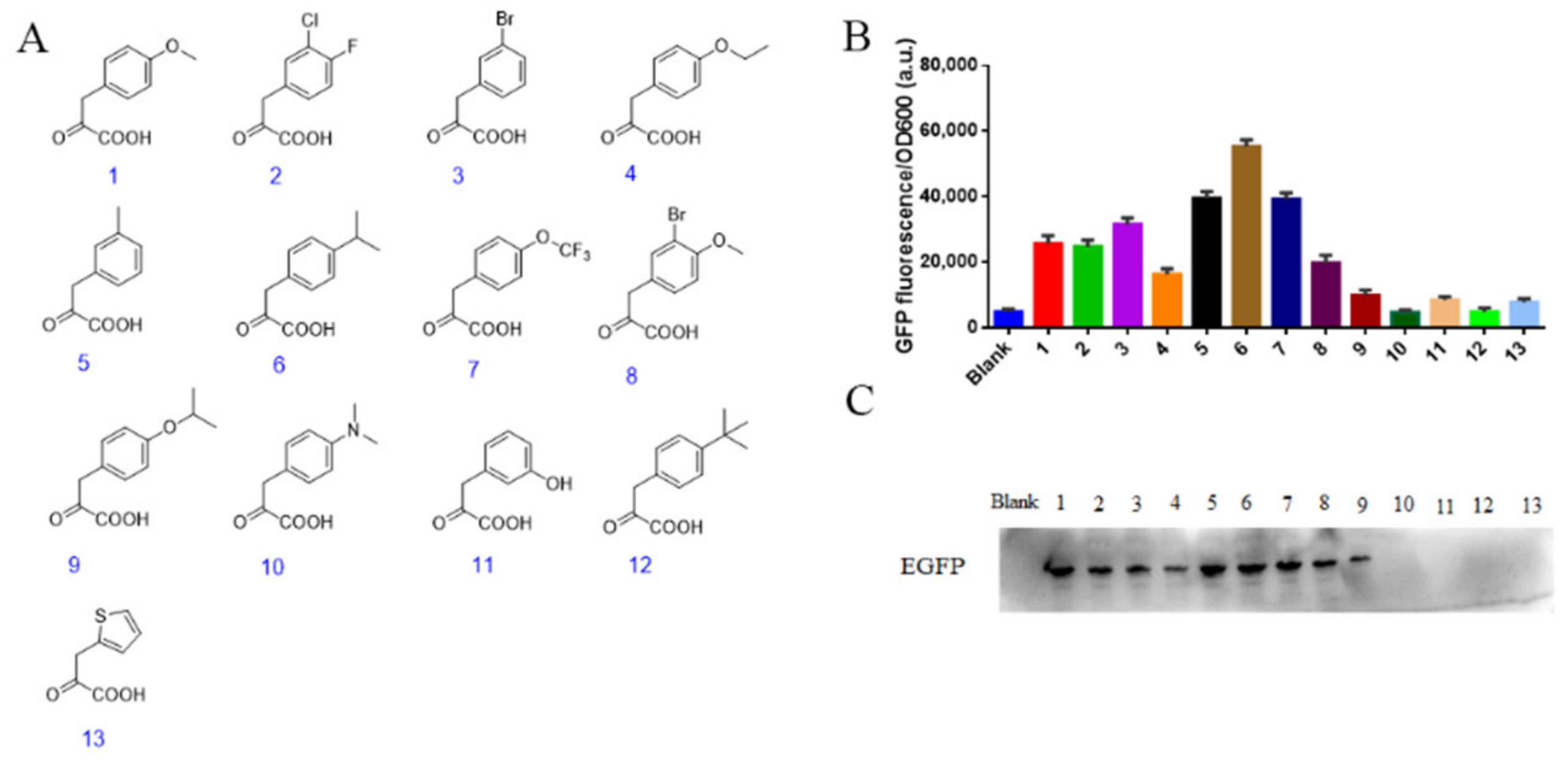
3.2. Screening for Bacterial Strains Utilizing the Keto-Acids
3.3. Effects of Substrate Concentration, Cofactor PLP and Amino Donor
3.4. Screening for Synthesised Keto Acids Introduction
3.5. The Evolution for GFPY66H/Y145X
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Chen, Q.; Klauser, P.C.; Li, M.; Zheng, F.; Wang, N.; Li, X.; Zhang, Q.; Fu, X.; Wang, Q.; et al. Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 2020, 182, 85–97.e16. [Google Scholar] [CrossRef]
- Wang, N.; Li, Y.; Niu, W.; Sun, M.; Cerny, R.; Li, Q.; Guo, J. Construction of a live-attenuated HIV-1 vaccine through genetic code expansion. Angew. Chemie Int. Ed. 2014, 53, 4867–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, A.J.; Lovelock, S.L.; Frese, A.; Crawshaw, R.; Ortmayer, M.; Dunstan, M.; Levy, C.; Green, A.P. Design and evolution of an enzyme with a non-canonical organocatalytic mechanism. Nature 2019, 570, 219–223. [Google Scholar] [CrossRef]
- Ling, X.; Xie, B.; Gao, X.; Chang, L.; Zheng, W.; Chen, H.; Huang, Y.; Tan, L.; Li, M.; Liu, T. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci. Adv. 2020, 6, eaaz0051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minks, C.; Huber, R.; Moroder, L.; Budisa, N. Noninvasive tracing of recombinant proteins with “Fluorophenylalanine-fingers”. Anal. Biochem. 2000, 284, 29–34. [Google Scholar] [CrossRef]
- Merkel, L.; Schauer, M.; Antranikian, G.; Budisa, N. Parallel incorporation of different fluorinated amino acids: On the way to “teflon” proteins. ChemBioChem 2010, 11, 1505–1507. [Google Scholar] [CrossRef]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ros, E.; Torres, A.G.; de Pouplana, L.R. Learning from Nature to Expand the Genetic Code. Trends Biotechnol. 2020, 39, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Martin, B.A.; King, D.S.; Wang, L.; Schultz, P.G. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Environ. Res. 1978, 15, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, W.; Schultz, P.G.; Guo, J. An expanded genetic code in mammalian cells with a functional quadruplet codon. ACS Chem. Biol. 2013, 8, 1640–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biava, H.D. Tackling Achilles’ Heel in Synthetic Biology: Pairing Intracellular Synthesis of Noncanonical Amino Acids with Genetic-Code Expansion to Foster Biotechnological Applications. ChemBioChem 2020, 21, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Giese, C.; Lepthien, S.; Metzner, L.; Brandsch, M.; Budisa, N.; Lilie, H. Intracellular uptake and inhibitory activity of aromatic fluorinated amino acids in human breast cancer cells. ChemMedChem 2008, 3, 1449–1456. [Google Scholar] [CrossRef]
- Völler, J.S.; Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr. Opin. Biotechnol. 2017, 48, 1–7. [Google Scholar] [CrossRef]
- Agostini, F.; Völler, J.S.; Koksch, B.; Acevedo-Rocha, C.G.; Kubyshkin, V.; Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew. Chemie Int. Ed. 2017, 56, 9680–9703. [Google Scholar] [CrossRef]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a bacterium with a 21 amino acid genetic code. J. Am. Chem. Soc. 2003, 125, 935–939. [Google Scholar] [CrossRef]
- Ou, W.; Uno, T.; Chiu, H.P.; Grünewald, J.; Cellitti, S.E.; Crossgrove, T.; Hao, X.; Fan, Q.; Quinn, L.L.; Patterson, P.; et al. Site-specific protein modifications through pyrroline-carboxy-lysine residues. Proc. Natl. Acad. Sci. USA 2011, 108, 10437–10442. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M.; Gattner, M.J.; Viverge, B.; Bretzler, J.; Eisen, D.; Stadlmeier, M.; Vrabel, M.; Carell, T. Orchestrating the biosynthesis of an unnatural pyrrolysine amino acid for its direct incorporation into proteins inside living cells. Chem. Eur. J. 2015, 21, 7701–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.E.; Lee, S.Y.; Park, H.; Cha, H.; Ko, W.; Sachin, K.; Kim, D.W.; Chi, D.Y.; Lee, H.S. Genetic incorporation of unnatural amino acids biosynthesized from α-keto acids by an aminotransferase. Chem. Sci. 2014, 5, 1881–1885. [Google Scholar] [CrossRef]
- Kim, S.; Sung, B.H.; Kim, S.C.; Lee, H.S. Genetic incorporation of l-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem. Commun. 2018, 54, 3002–3005. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.P.; Kuenzl, T.; To, T.M.T.; Ouyang, Z.; Schwagerus, S.; Hoesl, M.G.; Hackenberger, C.P.R.; Lensen, M.C.; Panke, S.; Budisa, N. Design of S-Allylcysteine in Situ Production and Incorporation Based on a Novel Pyrrolysyl-tRNA Synthetase Variant. ChemBioChem 2017, 18, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Nojoumi, S.; Ma, Y.; Schwagerus, S.; Hackenberger, C.P.R.; Budisa, N. In-cell synthesis of bioorthogonal alkene tag S-Allyl-homocysteine and its coupling with reprogrammed translation. Int. J. Mol. Sci. 2019, 20, 2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Di Salvo, M.L.; Budisa, N. Self-directed in cell production of methionine analogue azidohomoalanine by synthetic metabolism and its incorporation into model proteins. Methods Mol. Biol. 2018, 1728, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Biava, H.; Contestabile, R.; Budisa, N.; Di Salvo, M.L. Coupling bioorthogonal chemistries with artificial metabolism: Intracellular biosynthesis of azidohomoalanine and its incorporation into recombinant proteins. Molecules 2014, 19, 1004–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipp, C.J.; Ma, Y.; Al-Shameri, A.; D’Alessio, F.; Neubauer, P.; Contestabile, R.; Budisa, N.; di Salvo, M.L. An Engineered Escherichia coli Strain with Synthetic Metabolism for in-Cell Production of Translationally Active Methionine Derivatives. ChemBioChem 2020, 21, 3525–3538. [Google Scholar] [CrossRef]
- Schueler-Furman, O.; Altuvia, Y.; Margalit, H. Differential effects of isomeric incorporation of fluorophenylalanines into PvuII endonuclease. Proteins Struct. Funct. Genet. 2001, 45, 55–61. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Cai, W.; Tan, L.; Yu, Y.; Han, B.; Li, Y.; Xie, Y.; Su, Y.; Luo, X.; et al. Expanding the Structural Diversity of Protein Building Blocks with Noncanonical Amino Acids Biosynthesized from Aromatic Thiols. Angew. Chemie Int. Ed. 2021, 60, 10040–10048. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically Recoded Organisms Expand Biological Functions. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [Green Version]
- Hammerling, M.J.; Gollihar, J.; Mortensen, C.; Alnahhas, R.N.; Ellington, A.D.; Barrick, J.E. Expanded Genetic Codes Create New Mutational Routes to Rifampicin Resistance in Escherichia coli. Mol. Biol. Evol. 2016, 33, 2054–2063. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.T.; Lee, J.-I. Chemopreventive mechanisms of α-keto acid metabolites of naturally occurring organoselenium compounds. Amino Acids 2011, 41, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Deweid, L.; Avrutina, O.; Kolmar, H. Microbial transglutaminase for biotechnological and biomedical engineering. Biol. Chem. 2018, 400, 257–274. [Google Scholar] [CrossRef]
- Toney, M.D. Aspartate aminotransferase: An old dog teaches new tricks. Arch. Biochem. Biophys. 2014, 544, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girón, M.D.; Salto, R. From green to blue: Site-directed mutagenesis of the green fluorescent protein to teach protein structure-function relationships. Biochem. Mol. Biol. Educ. 2011, 39, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef] [PubMed]
- Craggs, T.D. Green fluorescent protein: Structure, folding and chromophore maturation. Chem. Soc. Rev. 2009, 38, 2865–2875. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Wang, B.; Li, S.; Xu, F.; He, Q.; Pan, C.; Gao, X.; Yao, W.; Song, X. Convenient Genetic Encoding of Phenylalanine Derivatives through Their α-Keto Acid Precursors. Biomolecules 2021, 11, 1358. https://doi.org/10.3390/biom11091358
Liu L, Wang B, Li S, Xu F, He Q, Pan C, Gao X, Yao W, Song X. Convenient Genetic Encoding of Phenylalanine Derivatives through Their α-Keto Acid Precursors. Biomolecules. 2021; 11(9):1358. https://doi.org/10.3390/biom11091358
Chicago/Turabian StyleLiu, Li, Bohao Wang, Sheng Li, Fengyuan Xu, Qi He, Chun Pan, Xiangdong Gao, Wenbing Yao, and Xiaoda Song. 2021. "Convenient Genetic Encoding of Phenylalanine Derivatives through Their α-Keto Acid Precursors" Biomolecules 11, no. 9: 1358. https://doi.org/10.3390/biom11091358