
Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases

 ,
,  , and
, and 
Abstract
: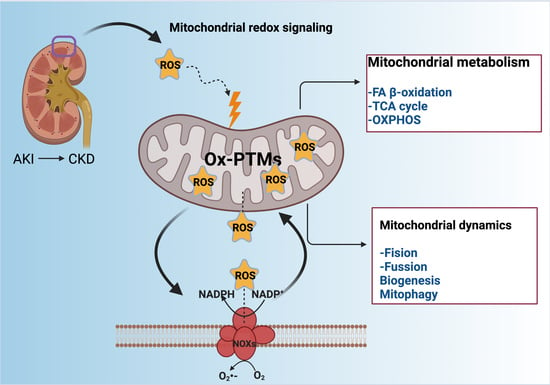
1. Introduction
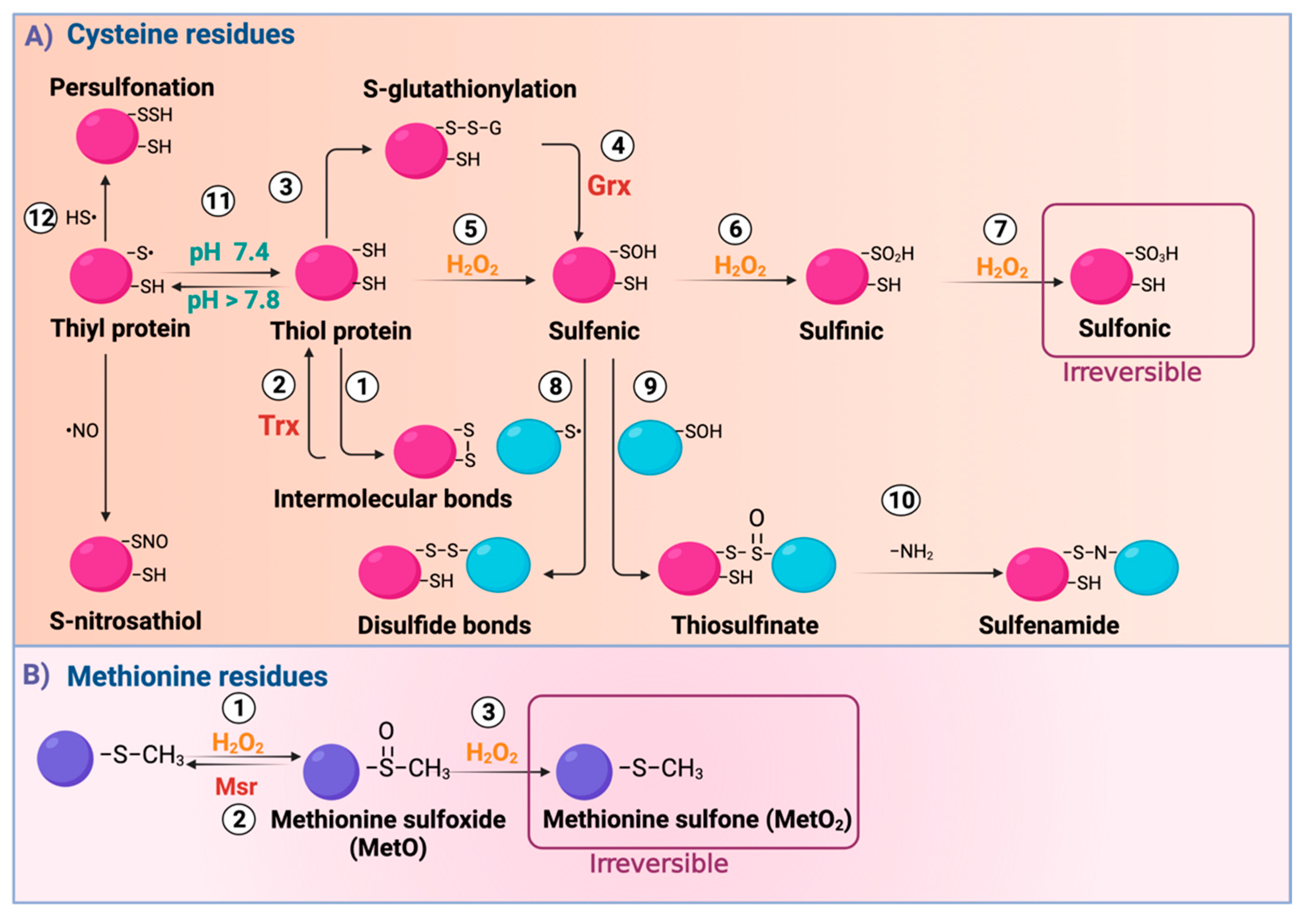
2. Redox-Sensitive Signaling in Kidney Diseases
3. Ox-PTMs Regulate Manganese Superoxide Dismutase (Mn-SOD) in Kidney Injury
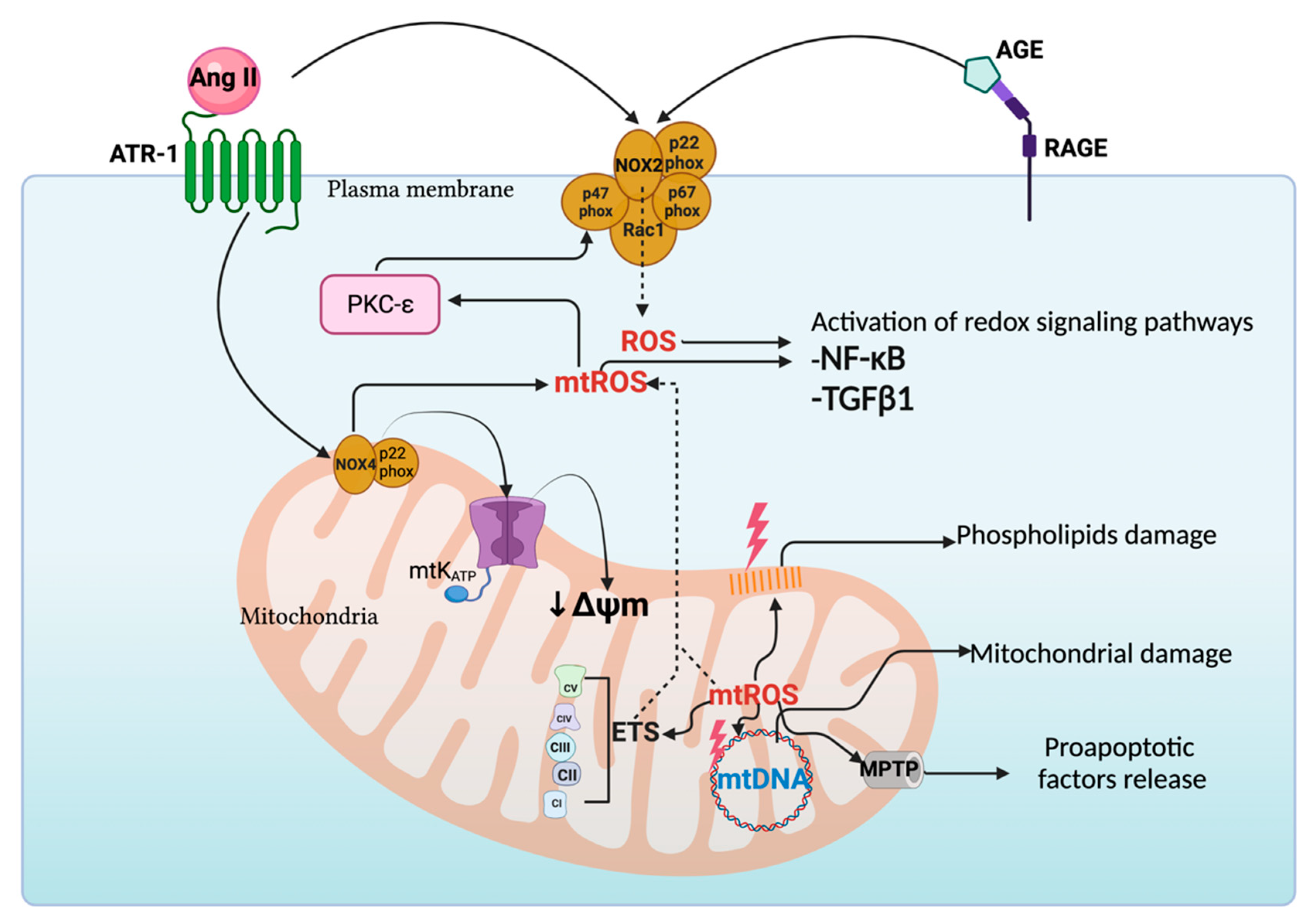
4. Crosstalk between NOXs and Mitochondria in Kidney Diseases
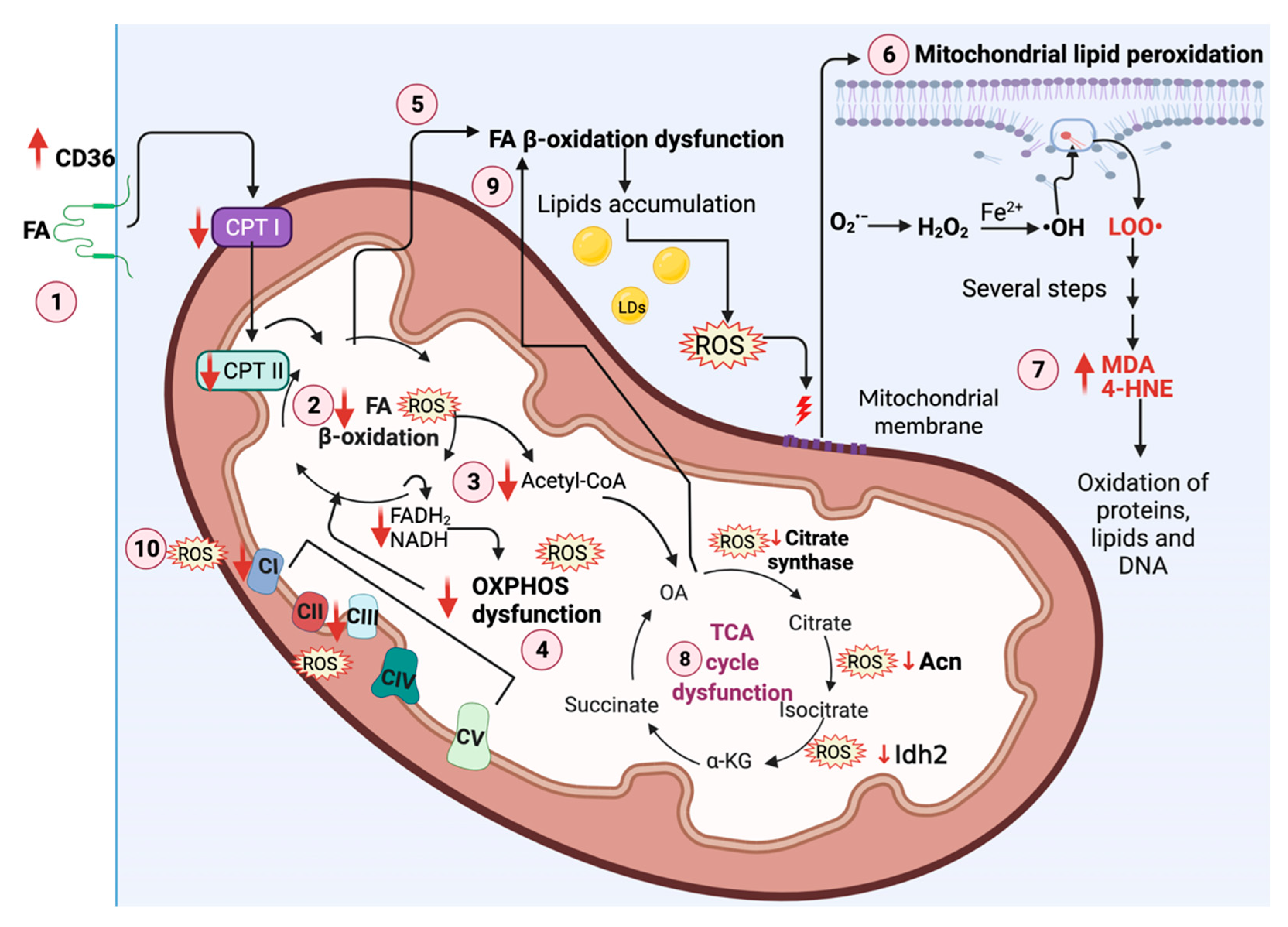
5. Mitochondrial Metabolism, ROS, and OS in Kidney Diseases
5.1. FA β-Oxidation Dysfunction in Kidney Diseases
Oxidation and OS Production and in Kidney Diseases
5.2. TCA Cycle Redox-Sensitive Signaling Pathway in Kidney Diseases
5.3. OXPHOS Redox-Sensitive Signaling Pathway in Kidney Diseases
6. ROS Induce Uncoupling Proteins (UCPs) Dysregulation in Kidney Diseases
7. Redox-Sensitive Signaling Controls Mitochondrial Dynamics, Biogenesis, and Mitophagy
7.1. Redox-Sensitive Proteins Participating in Fission and Fusion
7.2. Redox-Sensitive Proteins Participating in Mitochondrial Biogenesis
7.3. Mitophagy, ROS, and OS in Kidney Diseases
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ↓ΔΨm | mitochondrial membrane potential depolarization |
| •NO | nitric oxide |
| •OH | hydroxyl radical |
| 4-HNE | 4-hydroxynonenal |
| α-KΓ | Alpha-ketoglutarate |
| α-KΓΔH | alpha-ketoglutarate dehydrogenase |
| ACAD10 | acyl-CoA dehydrogenase family member 10 |
| ACS | acyl-CoA synthetase |
| Acn | aconitase |
| AGE | advanced glycation end products |
| AKR1A1 | aldo-keto reductase family 1 member A1 |
| AKI | acute kidney injury |
| AMPK | adenosine monophosphate (AMP)-activated protein kinase |
| Ang II | angiotensin II |
| Arg | arginine |
| ATP | adenosine triphosphate |
| ATR-1 | angiotensin type 1 receptor |
| Bcl-2 | B cell lymphoma 2 |
| BUN | blood urea nitrogen |
| CI | complex I |
| CII | complex II |
| CIII | complex III |
| CIV | complex IV |
| CV | complex V |
| C5aR1 | C5 substrate of the complement system receptor 1 |
| CAT | catalase |
| CBS | cystathionine β-synthase |
| CD36 | cluster of differentiation-36 |
| CKD | chronic kidney diseases |
| CoA | coenzyme A |
| Cox I | cytochrome c oxidase subunit 1 |
| CPT I | carnitine O-palmitoyl transferase I |
| CPT II | carnitine O-palmitoyl transferase II |
| CPT IA | CPT I isoform A |
| CPT IB | CPT I isoform B |
| CSE | cystathionine γ-lyase |
| Cu/Zn-SOD | copper/zinc-SOD |
| Cys | cysteine |
| CARSs | cysteinyl-transfer RNA (tRNA) synthetases |
| Cyt c | cytochrome c |
| DKD | diabetic kidney disease |
| DN | diabetic nephropathy |
| DNA | deoxyribonucleic acid |
| Drp1 | dynamin-related protein 1 |
| DT | distal tubules |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial–mesenchymal transition |
| eNOS | endothelial nitric oxide synthase |
| ERK2 | extracellular regulated kinase 2 |
| ERR | estrogen and estrogen-related receptors |
| ETS | electron transport system |
| FA | fatty acids |
| FABP | fatty-acid-binding protein |
| FADH2 | flavin adenine dinucleotide |
| Fe2+ | ferrous iron |
| Fe3+ | ferric iron |
| Fe-S | iron-sulfur |
| Fis1 | fission protein 1 |
| FoxO | forkhead box O |
| GC4419 | avasopasem manganese |
| GFRs | growth factor receptors |
| GPx | glutathione peroxidase |
| Grx | glutaredoxin |
| GSH | glutathione |
| GTPases | guanosine triphosphatases |
| GST | glutathione S-transferase |
| h | hours |
| HK2 | human proximal tubular cells |
| HS• | hydrosulfide radical. |
| H2O2 | hydrogen peroxide |
| H2S | gydrogen sulfide |
| His | histidine |
| I/R | ischemia/reperfusion |
| Idh2 | isocitrate dehydrogenase isoform 2 |
| IKKγ | regulatory subunit of inhibitor IKK complex |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| K2Cr2O7 | potassium dichromate |
| L-NAME | N(G)-nitro-L-arginine-methyl ester |
| LC3-II | microtubule-associated protein 1A/1B-light chain 3 phosphatidylethanolamine conjugate |
| LDs | lipid droplets |
| LOO• | lipid peroxyl radical |
| LPS | lipopolysaccharides |
| Lys | lysine |
| MA | maleic acid |
| MAPK | mitogen-activated protein kinases |
| MCAD | medium-chain acetyl dehydrogenase |
| MDA | malondialdehyde |
| Met | methionine |
| MetO (R–SOCH3) | methionine sulfoxide |
| MetO2 (RSO2CH3) | methionine sulfone |
| Mff | mitochondrial fission factor |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| Mief1 | mitochondrial elongation factor 1 |
| Mief2 | mitochondrial elongation factor 2 |
| mitoTEMPO | 2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl) triphenylphosphonium chloride |
| mitoQ | mitochondrial-targeting antioxidants coenzyme Q |
| Mn-SOD | manganese superoxide dismutase |
| mRNA | messenger RNA |
| Msr | methionine sulfoxide reductase |
| MsrA | Msr isoform A |
| MPTP | mitochondrial permeability transition pore |
| mt-KATP | mitochondrial ATP-sensitive potassium K channel |
| mtDNA | mitochondrial DNA |
| mtROS | mitochondrial ROS |
| Na+/K+ ATPase | sodium–potassium ATP pump |
| NAC | N-acetylcysteine |
| NAD | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NaHS | sodium hydrosulfide |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOS | nitric oxide synthase |
| NOXs | NADPH oxidases |
| NRF-1 | nuclear respiratory factor 1 |
| NRF-2 | nuclear respiratory factor 2 |
| Nrf2 | nuclear factor erythroid 2–related factor 2 |
| O2 | oxygen |
| O2•− | superoxide anion radical |
| OA | oxalacetate |
| OCR | oxygen consumption rate |
| OMM | outer mitochondrial membrane |
| ONOO− | peroxynitrite |
| Opa1 | optical atrophy 1 |
| OS | oxidative stress |
| Ox-PTMs | oxidative post-translational modifications |
| OXPHOS | oxidative phosphorylation |
| p62 | sequestosome |
| PDIA1 | protein disulfide isomerase A1 |
| PGC-1α | peroxisome proliferator-activated receptor (PPAR) γ coactivator-1 alpha |
| Pink1 | phosphatase and tensin homolog (PTEN)-induced putative kinase 1 |
| PKC | protein kinase C |
| PKC-ε | PKC-epsilon |
| PKC-δ | PKC-delta |
| PKM2 | pyruvate kinase isoform M2 |
| PMF | proton motive force |
| PPARs | peroxisome proliferator-activated receptor |
| Pro | proline |
| Prx | peroxiredoxin |
| PT | proximal tubules |
| PTKs | protein tyrosine kinases |
| PTM | post-translational modifications |
| PTPs | protein tyrosine phosphatases |
| R–S• | thyil radical |
| R–SNO | S-nitrosothiol |
| R–SOH | sulfenic acid |
| R–SO2H | sulfinic acid |
| R–SO3H | sulfonic acid |
| R–S(O)–S–R | thiosulfinate |
| R–SN–R’ | sulfenamide |
| R–S–S–H | persulfonation |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RAGE | AGE receptor |
| ROS | reactive oxygen species |
| RPTCs | renal proximal tubular cells |
| RTC | renal tubular cells |
| S− | thiolate anion |
| S–S | disulfide bonds |
| SF | sulforaphane |
| SH | thiol |
| siRNA | small interfering RNA |
| SIRT3 | nicotinamide adenine dinucleotide (NAD)-dependent deacetylase sirtuin-3 |
| SN2 | bimolecular nucleophilic substitution |
| SSG | glutathionylation |
| TCA | tricarboxylic acid |
| TECs | tubular epithelial cells |
| TFAM | transcription factor A |
| TGFβ1 | transforming growth factor-beta 1 |
| Thr | threonine |
| Trx | thioredoxin |
| Tyr | tyrosine |
| UCPs | uncoupling proteins |
| UCP1 | uncoupling protein 1 |
| UCP2 | uncoupling protein 2 |
| UCPs | uncoupling proteins |
| UUO | unilateral ureteral obstruction |
| VLCAD | very long-chain acyl-CoA dehydrogenase |
| WT | wild type |
References
- Manns, B.; Hemmelgarn, B.; Tonelli, M.; Au, F.; So, H.; Weaver, R.; Quinn, A.E.; Klarenbach, S.; for Canadians Seeking Solutions and Innovations to Overcome Chronic Kidney Disease. The Cost of Care for People With Chronic Kidney Disease. Can. J. Kidney Health Dis. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Collister, D.; Pannu, N.; Ye, F.; James, M.; Hemmelgarn, B.; Chui, B.; Manns, B.; Klarenbach, S. Health Care Costs Associated with AKI. Clin. J. Am. Soc. Nephrol. 2017, 12, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.E.; Blaine, C.; Dawnay, A.; Devonald, M.A.J.; Ftouh, S.; Laing, C.; Latchem, S.; Lewington, A.; Milford, D.V.; Ostermann, M. The Definition of Acute Kidney Injury and Its Use in Practice. Kidney Int. 2015, 87, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, Y.; Teng, J.; Ding, X. Acute Kidney Injury Epidemiology: From Recognition to Intervention. Contrib. Nephrol. 2016, 187, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic Kidney Disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Tapia, E.; Pedraza-Chaverri, J. Unilateral Ureteral Obstruction as a Model to Investigate Fibrosis-Attenuating Treatments. Biomolecules 2019, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Böttinger, E.P. Multiple Metabolic Hits Converge on CD36 as Novel Mediator of Tubular Epithelial Apoptosis in Diabetic Nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yuan, Q.; Xu, T.; Yao, L.; Feng, J.; Ma, J.; Wang, L.; Lu, C.; Wang, D. Pioglitazone Improves Mitochondrial Function in the Remnant Kidney and Protects against Renal Fibrosis in 5/6 Nephrectomized Rats. Front. Pharmacol. 2017, 8, 545. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Progression of Tubulointerstitial Fibrosis and the Chronic Kidney Disease Phenotype—Role of Risk Factors and Epigenetics. Front. Pharmacol. 2017, 8, 520. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial Energetics in the Kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms—Biomarkers—Interventions, and Future Perspectives. Oxidative Med. Cell. Longev. 2017, 2017, 6193694. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K. Redox-Sensitive Signalling Pathways Regulated by Human Papillomavirus in HPV-Related Cancers. Rev. Med. Virol. 2021, e2230. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Wani, R.; Nagata, A.; Murray, B.W. Protein Redox Chemistry: Post-Translational Cysteine Modifications That Regulate Signal Transduction and Drug Pharmacology. Front. Pharmacol. 2014, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein Carbonylation as a Novel Mechanism in Redox Signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Stamler, J.S. Enzymatic Mechanisms Regulating Protein S-Nitrosylation: Implications in Health and Disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Role of Metabolic H2O2 Generation: Redox Signaling and Oxidative Stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.C.; Ma, Y.S.; Stadtman, E.R. Modification of Protein Surface Hydrophobicity and Methionine Oxidation by Oxidative Systems. Proc. Natl. Acad. Sci. USA 1997, 94, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C.; Hampton, M.B. Thiol Chemistry and Specificity in Redox Signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox Regulation of Mitochondrial Function with Emphasis on Cysteine Oxidation Reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Conte, M.; Carroll, K.S. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, L.B.; Nelson, K.J. Discovering Mechanisms of Signaling-Mediated Cysteine Oxidation. Curr. Opin. Chem. Biol. 2008, 12, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, M.; Turell, L.; Manta, B.; Botti, H.; Monteiro, G.; Netto, L.E.S.; Alvarez, B.; Radi, R.; Trujillo, M. Thiol and Sulfenic Acid Oxidation of AhpE, the One-Cysteine Peroxiredoxin from Mycobacterium Tuberculosis: Kinetics, Acidity Constants, and Conformational Dynamics. Biochemistry 2009, 48, 9416–9426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.-W.; Singh, S.; Townsend, D.M.; Tew, K.D. An Evolving Understanding of the S-Glutathionylation Cycle in Pathways of Redox Regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Poole, L.B. The Basics of Thiols and Cysteines in Redox Biology and Chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Carroll, K.S. Sulfenic Acid Chemistry, Detection and Cellular Lifetime. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of Protein Function by Reversible Methionine Oxidation and the Role of Selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtman, E.R.; Moskovitz, J.; Levine, R.L. Oxidation of Methionine Residues of Proteins: Biological Consequences. Antioxid. Redox Signal. 2003, 5, 577–582. [Google Scholar] [CrossRef]
- García-Santamarina, S.; Boronat, S.; Hidalgo, E. Reversible Cysteine Oxidation in Hydrogen Peroxide Sensing and Signal Transduction. Biochemistry 2014, 53, 2560–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, A. Thioredoxin and Redox Signaling: Roles of the Thioredoxin System in Control of Cell Fate. Arch. Biochem. Biophys. 2017, 617, 101–105. [Google Scholar] [CrossRef]
- Roorda, M.; Miljkovic, J.L.; van Goor, H.; Henning, R.H.; Bouma, H.R. Spatiotemporal Regulation of Hydrogen Sulfide Signaling in the Kidney. Redox Biol. 2021, 43, 101961. [Google Scholar] [CrossRef]
- Sawa, T.; Motohashi, H.; Ihara, H.; Akaike, T. Enzymatic Regulation and Biological Functions of Reactive Cysteine Persulfides and Polysulfides. Biomolecules 2020, 10, 1245. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S Signalling through Protein Sulfhydration and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Winterbourn, C.C. Rapid Reaction of Hydrogen Sulfide with the Neutrophil Oxidant Hypochlorous Acid to Generate Polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef]
- Frank, G.D.; Eguchi, S. Activation of Tyrosine Kinases by Reactive Oxygen Species in Vascular Smooth Muscle Cells: Significance and Involvement of EGF Receptor Transactivation by Angiotensin II. Antioxid. Redox Signal. 2003, 5, 771–780. [Google Scholar] [CrossRef]
- Doulias, P.-T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric Oxide Regulates Mitochondrial Fatty Acid Metabolism Through Reversible Protein S-Nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic Reprogramming by the S-Nitroso-CoA Reductase System Protects against Kidney Injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Siow, Y.L.; Isaak, C.K.; O, K. Downregulation of Glutathione Biosynthesis Contributes to Oxidative Stress and Liver Dysfunction in Acute Kidney Injury. Available online: https://www.hindawi.com/journals/omcl/2016/9707292/ (accessed on 5 February 2021).
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective Effects of N-Acetyl-Cysteine in Mitochondria Bioenergetics, Oxidative Stress, Dynamics and S-Glutathionylation Alterations in Acute Kidney Damage Induced by Folic Acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Noh, M.R.; Kim, K.Y.; Han, S.J.; Kim, J.I.; Kim, H.-Y.; Park, K.M. Methionine Sulfoxide Reductase A Deficiency Exacerbates Cisplatin-Induced Nephrotoxicity via Increased Mitochondrial Damage and Renal Cell Death. Antioxid. Redox Signal. 2017, 27, 727–741. [Google Scholar] [CrossRef]
- Kim, J.I.; Choi, S.H.; Jung, K.-J.; Lee, E.; Kim, H.-Y.; Park, K.M. Protective Role of Methionine Sulfoxide Reductase A against Ischemia/Reperfusion Injury in Mouse Kidney and Its Involvement in the Regulation of Trans-Sulfuration Pathway. Antioxid. Redox Signal. 2013, 18, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.-J.; Jang, H.-S.; Kim, J.I.; Han, S.J.; Park, J.-W.; Park, K.M. Involvement of Hydrogen Sulfide and Homocysteine Transsulfuration Pathway in the Progression of Kidney Fibrosis after Ureteral Obstruction. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1989–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.I.; Noh, M.R.; Kim, K.Y.; Jang, H.-S.; Kim, H.-Y.; Park, K.M. Methionine Sulfoxide Reductase A Deficiency Exacerbates Progression of Kidney Fibrosis Induced by Unilateral Ureteral Obstruction. Free Radic. Biol. Med. 2015, 89, 201–208. [Google Scholar] [CrossRef]
- Lai, L.; Sun, J.; Tarafdar, S.; Liu, C.; Murphy, E.; Kim, G.; Levine, R.L. Loss of Methionine Sulfoxide Reductases Increases Resistance to Oxidative Stress. Free Radic. Biol. Med. 2019, 145, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Bian, J.-S. The Role of Hydrogen Sulfide in Renal System. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasinath, B.S.; Feliers, D.; Lee, H.J. Hydrogen Sulfide as a Regulatory Factor in Kidney Health and Disease. Biochem. Pharmacol. 2018, 149, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ngowi, E.E.; Sarfraz, M.; Afzal, A.; Khan, N.H.; Khattak, S.; Zhang, X.; Li, T.; Duan, S.-F.; Ji, X.-Y.; Wu, D.-D. Roles of Hydrogen Sulfide Donors in Common Kidney Diseases. Front. Pharmacol. 2020, 11, 1706. [Google Scholar] [CrossRef]
- Han, S.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Hydrogen Sulfide Accelerates the Recovery of Kidney Tubules after Renal Ischemia/Reperfusion Injury. Nephrol. Dial. Transplant. 2015, 30, 1497–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.-L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine γ-Lyase Protects against Renal Ischemia/Reperfusion by Modulating Oxidative Stress. J. Am. Soc. Nephrol. 2013, 24, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Feng, S.-J.; Zhang, G.-Z.; Wang, S.-X. Correlation of Lower Concentrations of Hydrogen Sulfide with Atherosclerosis in Chronic Hemodialysis Patients with Diabetic Nephropathy. Blood Purif. 2014, 38, 188–194. [Google Scholar] [CrossRef]
- Song, K.; Wang, F.; Li, Q.; Shi, Y.-B.; Zheng, H.-F.; Peng, H.; Shen, H.-Y.; Liu, C.-F.; Hu, L.-F. Hydrogen Sulfide Inhibits the Renal Fibrosis of Obstructive Nephropathy. Kidney Int. 2014, 85, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S Signals through Protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarosz, A.P.; Wei, W.; Gauld, J.W.; Auld, J.; Özcan, F.; Aslan, M.; Mutus, B. Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH) Is Inactivated by S-Sulfuration in Vitro. Free Radic. Biol. Med. 2015, 89, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 Defines a Physiologically Important Stress Response Mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Bianco, M.; Lopes, J.A.; Beiral, H.J.V.; Filho, J.D.D.; Frankenfeld, S.P.; Fortunato, R.S.; Gattass, C.R.; Vieyra, A.; Takiya, C.M. The Contralateral Kidney Presents with Impaired Mitochondrial Functions and Disrupted Redox Homeostasis after 14 Days of Unilateral Ureteral Obstruction in Mice. PLoS ONE 2019, 14, e0218986. [Google Scholar] [CrossRef]
- Kong, W.; Fu, J.; Liu, N.; Jiao, C.; Guo, G.; Luan, J.; Wang, H.; Yao, L.; Wang, L.; Yamamoto, M.; et al. Nrf2 Deficiency Promotes the Progression from Acute Tubular Damage to Chronic Renal Fibrosis Following Unilateral Ureteral Obstruction. Nephrol. Dial. Transplant. 2018, 33, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.-N.; Zhao, M.-M.; Wu, D.-D.; Chen, Y.; Wang, Y.; Zhu, J.-H.; Cai, W.-J.; Zhu, Y.-Z.; Zhu, Y.-C. Hydrogen Sulfide Targets EGFR Cys797/Cys798 Residues to Induce Na+/K+-ATPase Endocytosis and Inhibition in Renal Tubular Epithelial Cells and Increase Sodium Excretion in Chronic Salt-Loaded Rats. Antioxid. Redox Signal. 2014, 21, 2061–2082. [Google Scholar] [CrossRef] [Green Version]
- Shirozu, K.; Tokuda, K.; Marutani, E.; Lefer, D.; Wang, R.; Ichinose, F. Cystathionine γ-Lyase Deficiency Protects Mice from Galactosamine/Lipopolysaccharide-Induced Acute Liver Failure. Antioxid. Redox Signal. 2014, 20, 204–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, P.K.; Martinov, M.; Vitvitsky, V.; Seravalli, J.; Wedmann, R.; Filipovic, M.R.; Banerjee, R. Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J. Am. Chem. Soc. 2016, 138, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, S.; Ishii, I.; Shinmura, K.; Tamaki, K.; Hishiki, T.; Akahoshi, N.; Ida, T.; Nakanishi, T.; Kamata, S.; Kumagai, Y.; et al. Hyperhomocysteinemia Abrogates Fasting-Induced Cardioprotection against Ischemia/Reperfusion by Limiting Bioavailability of Hydrogen Sulfide Anions. J. Mol. Med. 2015, 93, 879–889. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-TRNA Synthetase Governs Cysteine Polysulfidation and Mitochondrial Bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnati, S.; Lüers, G.; Pfreimer, S.; Baumgart-Vogt, E. Mammalian SOD2 Is Exclusively Located in Mitochondria and Not Present in Peroxisomes. Histochem. Cell Biol. 2013, 140, 105–117. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Yamakura, F.; Kawasaki, H. Post-Translational Modifications of Superoxide Dismutase. Biochim. Biophys. Acta 2010, 1804, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Walker, P.D.; Imam, S.Z.; Ali, S.F.; Mayeux, P.R. Evidence for Peroxynitrite Formation in Renal Ischemia-Reperfusion Injury: Studies with the Inducible Nitric Oxide Synthase Inhibitor L-N(6)-(1-Iminoethyl)Lysine. J. Pharmacol. Exp. Ther. 2000, 295, 417–422. [Google Scholar] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Kerby, J.D.; Beckman, J.S.; Thompson, J.A. Nitration and Inactivation of Manganese Superoxide Dismutase in Chronic Rejection of Human Renal Allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar] [CrossRef] [Green Version]
- MacMillan-Crow, L.A.; Cruthirds, D.L.; Ahki, K.M.; Sanders, P.W.; Thompson, J.A. Mitochondrial Tyrosine Nitration Precedes Chronic Allograft Nephropathy. Free Radic. Biol. Med. 2001, 31, 1603–1608. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-Mediated Inactivation of Manganese Superoxide Dismutase Involves Nitration and Oxidation of Critical Tyrosine Residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef]
- Cruthirds, D.L.; Novak, L.; Akhi, K.M.; Sanders, P.W.; Thompson, J.A.; MacMillan-Crow, L.A. Mitochondrial Targets of Oxidative Stress during Renal Ischemia/Reperfusion. Arch. Biochem. Biophys. 2003, 412, 27–33. [Google Scholar] [CrossRef]
- Xu, S.; Jiang, B.; Maitland, K.A.; Bayat, H.; Gu, J.; Nadler, J.L.; Corda, S.; Lavielle, G.; Verbeuren, T.J.; Zuccollo, A.; et al. The Thromboxane Receptor Antagonist S18886 Attenuates Renal Oxidant Stress and Proteinuria in Diabetic Apolipoprotein E-Deficient Mice. Diabetes 2006, 55, 110–119. [Google Scholar] [CrossRef]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH Oxidase in the Vascular Hypertrophic and Oxidative Stress Response to Angiotensin II in Mice. Circ. Res. 2001, 88, 947–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Adachi, T.; Matsui, R.; Xu, S.; Jiang, B.; Zou, M.-H.; Kirber, M.; Lieberthal, W.; Cohen, R.A. Quantitative Assessment of Tyrosine Nitration of Manganese Superoxide Dismutase in Angiotensin II-Infused Rat Kidney. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1396–H1403. [Google Scholar] [CrossRef] [Green Version]
- Majzunova, M.; Kvandova, M.; Berenyiova, A.; Balis, P.; Dovinova, I.; Cacanyiova, S. Chronic NOS Inhibition Affects Oxidative State and Antioxidant Response Differently in the Kidneys of Young Normotensive and Hypertensive Rats. Oxidative Med. Cell. Longev. 2019, 2019, 5349398. [Google Scholar] [CrossRef]
- Kimura, T.; Shiizaki, K.; Akimoto, T.; Shinzato, T.; Shimizu, T.; Kurosawa, A.; Kubo, T.; Nanmoku, K.; Kuro-O, M.; Yagisawa, T. The Impact of Preserved Klotho Gene Expression on Antioxidative Stress Activity in Healthy Kidney. Am. J. Physiol. Physiol. 2018, 315, F345–F352. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Babelova, A.; Avaniadi, D.; Jung, O.; Fork, C.; Beckmann, J.; Kosowski, J.; Weissmann, N.; Anilkumar, N.; Shah, A.M.; Schaefer, L.; et al. Role of Nox4 in Murine Models of Kidney Disease. Free Radic. Biol. Med. 2012, 53, 842–853. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 Activity Is Determined by MRNA Levels and Reveals a Unique Pattern of ROS Generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Jose, P.A. Coordinated Contribution of NADPH Oxidase- and Mitochondria-Derived Reactive Oxygen Species in Metabolic Syndrome and Its Implication in Renal Dysfunction. Front. Pharmacol. 2021, 12, 670076. [Google Scholar] [CrossRef]
- Molina-Jijón, E.; Aparicio-Trejo, O.E.; Rodríguez-Muñoz, R.; León-Contreras, J.C.; Cárdenas-Aguayo, M.d.C.; Medina-Campos, O.N.; Tapia, E.; Sánchez-Lozada, L.G.; Hernández-Pando, R.; Reyes, J.L.; et al. The Nephroprotection Exerted by Curcumin in Maleate-Induced Renal Damage Is Associated with Decreased Mitochondrial Fission and Autophagy: Curcumin Decreases Maleate-Induced Renal Dysfunction. BioFactors 2016, 42, 686–702. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.J.; Yoon, H.E.; Chung, S.; Choi, B.S.; Park, C.W.; Shin, S.J. Fimasartan, a Novel Angiotensin-Receptor Blocker, Protects against Renal Inflammation and Fibrosis in Mice with Unilateral Ureteral Obstruction: The Possible Role of Nrf2. Int. J. Med. Sci. 2015, 12, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S. Cross Talk between Mitochondria and NADPH Oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.D.; Müller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First Evidence for a Crosstalk Between Mitochondrial and NADPH Oxidase-Derived Reactive Oxygen Species in Nitroglycerin-Triggered Vascular Dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A. Redox Signaling (Cross-Talk) from and to Mitochondria Involves Mitochondrial Pores and Reactive Oxygen Species. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallan, S.; Sharma, K. The Role of Mitochondria in Diabetic Kidney Disease. Curr. Diabetes Rep. 2016, 16, 61. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia Activates NADPH Oxidase to Increase [ROS]i and [Ca2+]i through Mitochondrial ROS–PKCε Signaling Axis in Pulmonary Artery Smooth Muscle Cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Mitochondrial Bioenergetics, Redox State, Dynamics and Turnover Alterations in Renal Mass Reduction Models of Chronic Kidney Diseases and Their Possible Implications in the Progression of This Illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef]
- Cheng, X.; Zheng, X.; Song, Y.; Qu, L.; Tang, J.; Meng, L.; Wang, Y. Apocynin Attenuates Renal Fibrosis via Inhibition of NOXs-ROS-ERK-Myofibroblast Accumulation in UUO Rats. Free Radic. Res. 2016, 50, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH Oxidase Inhibitor in Diabetic Nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Ortega-Lozano, A.J.; Pedraza-Chaverri, J. Redox Signaling Pathways in Unilateral Ureteral Obstruction (UUO)-Induced Renal Fibrosis. Free Radic. Biol. Med. 2021, 172, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, N.; Tamada, S.; Iwai, T.; Teramoto, K.; Kaneda, N.; Yukimura, T.; Nakatani, T.; Miura, K. Attenuation of Renal Fibrosis by Curcumin in Rat Obstructive Nephropathy. Urology 2006, 67, 440–446. [Google Scholar] [CrossRef]
- Herb, M.; Gluschko, A.; Wiegmann, K.; Farid, A.; Wolf, A.; Utermöhlen, O.; Krut, O.; Krönke, M.; Schramm, M. Mitochondrial Reactive Oxygen Species Enable Proinflammatory Signaling through Disulfide Linkage of NEMO. Sci. Signal. 2019, 12, eaar5926. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.B.Y.; Chen, W.; Fan, J.; Li, S.; Qu, X.; Chen, Q.; Chen, R.; Zhu, D.; Zhang, J.; et al. Smad4 Promotes Diabetic Nephropathy by Modulating Glycolysis and OXPHOS. EMBO Rep. 2020, 21, e48781. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Zuryn, S. Mitochondrial Genome (MtDNA) Mutations That Generate Reactive Oxygen Species. Antioxidants 2019, 8, 392. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regener. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Renal Glucose Metabolism in Normal Physiological Conditions and in Diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Pfaller, W.; Rittinger, M. Quantitative Morphology of the Rat Kidney. Int. J. Biochem. 1980, 12, 17–22. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markwell, M.A.; McGroarty, E.J.; Bieber, L.L.; Tolbert, N.E. The Subcellular Distribution of Carnitine Acyltransferases in Mammalian Liver and Kidney. A New Peroxisomal Enzyme. J. Biol. Chem. 1973, 248, 3426–3432. [Google Scholar] [CrossRef]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The Evolving Understanding of the Contribution of Lipid Metabolism to Diabetic Kidney Disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective Fatty Acid Oxidation in Renal Tubular Epithelial Cells Has a Key Role in Kidney Fibrosis Development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, L.V.; Tamirisa, A.; Kennedy, D.J.; Haller, S.T.; Budnyy, G.; Shapiro, J.I.; Malhotra, D. Mitochondrial Impairment in the Five-Sixth Nephrectomy Model of Chronic Renal Failure: Proteomic Approach. BMC Nephrol. 2013, 14, 209. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Chronic Impairment of Mitochondrial Bioenergetics and β-Oxidation Promotes Experimental AKI-to-CKD Transition Induced by Folic Acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Briones-Herrera, A.; Ramírez-Camacho, I.; Zazueta, C.; Tapia, E.; Pedraza-Chaverri, J. Altered Proximal Tubule Fatty Acid Utilization, Mitophagy, Fission and Supercomplexes Arrangement in Experimental Fanconi Syndrome Are Ameliorated by Sulforaphane-Induced Mitochondrial Biogenesis. Free Radic. Biol. Med. 2020, 153, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Ziemann, M.; Thallas-Bonke, V.; Snelson, M.; Kumar, V.; Laskowski, A.; Nguyen, T.-V.; Huynh, K.; Clarke, M.V.; Libianto, R.; et al. Complement C5a Induces Renal Injury in Diabetic Kidney Disease by Disrupting Mitochondrial Metabolic Agility. Diabetes 2020, 69, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512. [Google Scholar] [CrossRef]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered Renal Lipid Metabolism and Renal Lipid Accumulation in Human Diabetic Nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Declèves, A.-E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of Lipid Accumulation by AMP-Activated Kinase [Corrected] in High Fat Diet-Induced Kidney Injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, S.M.S.; Ashtiyani, S.C.; Hosseinkhani, S.; Shirazi, M. Comparison of the Effects of L: -Carnitine and Alpha-Tocopherol on Acute Ureteral Obstruction-Induced Renal Oxidative Imbalance and Altered Energy Metabolism in Rats. Urol. Res. 2010, 38, 187–194. [Google Scholar] [CrossRef]
- Bobulescu, I.A. Renal Lipid Metabolism and Lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Okamura, D.M.; Pennathur, S.; Pasichnyk, K.; López-Guisa, J.M.; Collins, S.; Febbraio, M.; Heinecke, J.; Eddy, A.A. CD36 Regulates Oxidative Stress and Inflammation in Hypercholesterolemic CKD. J. Am. Soc. Nephrol. 2009, 20, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-J.; Ghosh, S.; Kovalik, J.-P.; Ching, J.; Choi, H.W.; Tavintharan, S.; Ong, C.N.; Sum, C.F.; Summers, S.A.; Tai, E.S.; et al. Profiling of Plasma Metabolites Suggests Altered Mitochondrial Fuel Usage and Remodeling of Sphingolipid Metabolism in Individuals With Type 2 Diabetes and Kidney Disease. Kidney Int. Rep. 2017, 2, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Carrasco, R.; García-Arroyo, F.E.; Aparicio-Trejo, O.E.; Rojas-Morales, P.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Tapia, E.; Pedraza-Chaverri, J. Progressive Reduction in Mitochondrial Mass Is Triggered by Alterations in Mitochondrial Biogenesis and Dynamics in Chronic Kidney Disease Induced by 5/6 Nephrectomy. Biology 2021, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal Tubule Cpt1a Overexpression Protects from Kidney Fibrosis by Restoring Mitochondrial Homeostasis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Kakimoto, P.A.H.B.; Kowaltowski, A.J. Diet-Sensitive Sources of Reactive Oxygen Species in Liver Mitochondria: Role of Very Long Chain Acyl-CoA Dehydrogenases. PLoS ONE 2013, 8, e77088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimoto, P.A.H.B.; Tamaki, F.K.; Cardoso, A.R.; Marana, S.R.; Kowaltowski, A.J. H2O2 Release from the Very Long Chain Acyl-CoA Dehydrogenase. Redox Biol. 2015, 4, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walport, M.J. Complement. Available online: https://www.nejm.org/doi/10.1056/NEJM200104053441406 (accessed on 18 June 2021).
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR Activation Attenuate Cisplatin Induced AKI by Inhibiting Ferroptosis. Cell Death Dis. 2020, 11, 73. [Google Scholar] [CrossRef]
- Deng, B.; Yang, W.; Wang, D.; Cheng, L.; Bu, L.; Rao, J.; Zhang, J.; Xie, J.; Zhang, B. Peptide DR8 Suppresses Epithelial-to-Mesenchymal Transition via the TGF-β/MAPK Signaling Pathway in Renal Fibrosis. Life Sci. 2020, 261, 118465. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin Prevents Mitochondrial Dynamics Disturbances in Early 5/6 Nephrectomy: Relation to Oxidative Stress and Mitochondrial Bioenergetics. Biofactors 2017, 43, 293–310. [Google Scholar] [CrossRef]
- Ge, M.; Fontanesi, F.; Merscher, S.; Fornoni, A. The Vicious Cycle of Renal Lipotoxicity and Mitochondrial Dysfunction. Front. Physiol. 2020, 11, 732. [Google Scholar] [CrossRef]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Gómez-Sierra, T.; Jiménez-Uribe, A.P.; Bellido, B.; Pedraza-Chaverri, J. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in the Promotion of Fibrosis in Obstructive Nephropathy Induced by Unilateral Ureteral Obstruction. BioFactors 2020, 46, 716–733. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Manzo-Merino, J.; Lizano, M. Cellular Redox, Cancer and Human Papillomavirus. Virus Res. 2018, 246, 35–45. [Google Scholar] [CrossRef]
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, Ľ. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxidative Med. Cell. Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; Wei, W.; Fan, X.; Ci, X. Farrerol Attenuates Cisplatin-Induced Nephrotoxicity by Inhibiting the Reactive Oxygen Species-Mediated Oxidation, Inflammation, and Apoptotic Signaling Pathways. Front. Physiol. 2019, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.A.G.; Catão, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.P.; Santos, A.C. Cisplatin-Induced Nephrotoxicity Is Associated with Oxidative Stress, Redox State Unbalance, Impairment of Energetic Metabolism and Apoptosis in Rat Kidney Mitochondria. Arch. Toxicol. 2007, 81, 495–504. [Google Scholar] [CrossRef]
- Kaeidi, A.; Taghipour, Z.; Allahtavakoli, M.; Fatemi, I.; Hakimizadeh, E.; Hassanshahi, J. Ameliorating Effect of Troxerutin in Unilateral Ureteral Obstruction Induced Renal Oxidative Stress, Inflammation, and Apoptosis in Male Rats. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 879–888. [Google Scholar] [CrossRef]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-Regulation of the Citric Acid (TCA) Cycle in Non-Diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Hyeon, J.S.; Kim, N.H.; Cho, A.; Lee, G.; Jang, S.Y.; Kim, M.-K.; Lee, E.Y.; Chung, C.H.; Ha, H.; et al. Metabolic Changes in Urine and Serum during Progression of Diabetic Kidney Disease in a Mouse Model. Arch. Biochem. Biophys. 2018, 646, 90–97. [Google Scholar] [CrossRef]
- Liu, H.; Li, W.; He, Q.; Xue, J.; Wang, J.; Xiong, C.; Pu, X.; Nie, Z. Mass Spectrometry Imaging of Kidney Tissue Sections of Rat Subjected to Unilateral Ureteral Obstruction. Sci. Rep. 2017, 7, 41954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.-K.; Harris, R.C. EGF Receptor Deletion in Podocytes Attenuates Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Dugan, L.L.; You, Y.-H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.-E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK Dysregulation Promotes Diabetes-Related Reduction of Superoxide and Mitochondrial Function. Available online: https://www.jci.org/articles/view/66218/pdf (accessed on 17 June 2021).
- Mailloux, R.J.; Hamel, R.; Appanna, V.D. Aluminum Toxicity Elicits a Dysfunctional TCA Cycle and Succinate Accumulation in Hepatocytes. J. Biochem. Mol. Toxicol. 2006, 20, 198–208. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent Increase in Mitochondrial Superoxide Mediates Cisplatin-Induced Chronic Kidney Disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Jang, H.-S.; Noh, M.R.; Kim, J.; Kong, M.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase Deficiency Exacerbates Mitochondrial and Cell Damage after Kidney Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1200–1215. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.J.; Han, S.J.; Kim, J.I.; Park, J.-W.; Park, K.M. Mitochondrial NADP + -Dependent Isocitrate Dehydrogenase Deficiency Increases Cisplatin-Induced Oxidative Damage in the Kidney Tubule Cells. Cell Death Dis. 2018, 9, 716. [Google Scholar] [CrossRef]
- Kil, I.S.; Park, J.-W. Regulation of Mitochondrial NADP+-Dependent Isocitrate Dehydrogenase Activity by Glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Noh, M.R.; Yoon, G.-E.; Jang, H.-S.; Kong, M.J.; Park, K.M. IDH2 Gene Deficiency Accelerates Unilateral Ureteral Obstruction-Induced Kidney Inflammation through Oxidative Stress and Activation of Macrophages. Korean J. Physiol. Pharmacol. 2021, 25, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Briones-Herrera, A.; Medina-Campos, O.N.; Reyes-Fermín, L.M.; Martínez-Klimova, E.; León-Contreras, J.C.; Hernández-Pando, R.; Tapia, E.; Pedraza-Chaverri, J. Alterations in Mitochondrial Homeostasis in a Potassium Dichromate Model of Acute Kidney Injury and Their Mitigation by Curcumin. Food Chem. Toxicol. 2020, 145, 111774. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.-E. Uncoupling Proteins and the Control of Mitochondrial Reactive Oxygen Species Production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cai, T.; Xu, J.; Jiang, L.; Wu, J.; Sun, Q.; Zen, K.; Yang, J. UCP2 Attenuates Apoptosis of Tubular Epithelial Cells in Renal Ischemia-Reperfusion Injury. Am. J. Physiol. Physiol. 2017, 313, F926–F937. [Google Scholar] [CrossRef]
- Zhong, X.; He, J.; Zhang, X.; Li, C.; Tian, X.; Xia, W.; Gan, H.; Xia, Y. UCP2 Alleviates Tubular Epithelial Cell Apoptosis in Lipopolysaccharide-Induced Acute Kidney Injury by Decreasing ROS Production. Biomed. Pharmacother. 2019, 115, 108914. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Qiu, W.; Zhou, Y.; Wen, P.; Fang, L.; Cao, H.; Zen, K.; He, W.; Zhang, C.; Dai, C.; et al. A MicroRNA-30e/Mitochondrial Uncoupling Protein 2 Axis Mediates TGF-Β1-Induced Tubular Epithelial Cell Extracellular Matrix Production and Kidney Fibrosis. Kidney Int. 2013, 84, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling Protein 1 Inhibits Mitochondrial Reactive Oxygen Species Generation and Alleviates Acute Kidney Injury. EBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS Regulate Thermogenic Energy Expenditure and Sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The Intracellular Redox State Is a Core Determinant of Mitochondrial Fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Disatnik, M.-H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant Mitochondrial Fission in Neurons Induced by Protein Kinase C{delta} under Oxidative Stress Conditions in Vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Youn, S.-W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; León-Contreras, J.C.; Granados-Pineda, J.; Hernández-Pando, R.; Gonzaga, G.; Sánchez-Lozada, L.G.; Osorio-Alonso, H.; Pedraza-Chaverri, J.; Tapia, E. Protection against Renal Ischemia and Reperfusion Injury by Short-Term Time-Restricted Feeding Involves the Mitochondrial Unfolded Protein Response. Free Radic. Biol. Med. 2020, 154, 75–83. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.-G.; Dong, Z. Regulation of Mitochondrial Dynamics in Acute Kidney Injury in Cell Culture and Rodent Models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Ortega-Domínguez, B.; Aparicio-Trejo, O.E.; García-Arroyo, F.E.; León-Contreras, J.C.; Tapia, E.; Molina-Jijón, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Barrera-Oviedo, D.; Pedraza-Chaverri, J. Curcumin Prevents Cisplatin-Induced Renal Alterations in Mitochondrial Bioenergetics and Dynamic. Food Chem. Toxicol. 2017, 107, 373–385. [Google Scholar] [CrossRef]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial Dysfunction in Kidney Injury, Inflammation, and Disease: Potential Therapeutic Approaches. Kidney Res. Clin. Pract. 2020, 39, 244–258. [Google Scholar] [CrossRef]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-Regulated PARK2-Dependent Mitophagy Protects against Renal Fibrosis in Unilateral Ureteral Obstruction. Free Radic. Biol. Med. 2020, 152, 632–649. [Google Scholar] [CrossRef]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Yang, Q.; Zhang, X.; Qin, R.; Shan, W.; Zhang, H.; Chen, X. Quercetin Alleviates Kidney Fibrosis by Reducing Renal Tubular Epithelial Cell Senescence through the SIRT1/PINK1/Mitophagy Axis. Life Sci. 2020, 257, 118116. [Google Scholar] [CrossRef]
- Sang, X.-Y.; Xiao, J.-J.; Liu, Q.; Zhu, R.; Dai, J.-J.; Zhang, C.; Yu, H.; Yang, S.-J.; Zhang, B.-F. Regulators of Calcineurin 1 Deficiency Attenuates Tubulointerstitial Fibrosis through Improving Mitochondrial Fitness. FASEB J. 2020, 34. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric Oxide-Induced Mitochondrial Fission Is Regulated by Dynamin-Related GTPases in Neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Villena, J.A. New Insights into PGC-1 Coactivators: Redefining Their Role in the Regulation of Mitochondrial Function and Beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Hock, M.B.; Kralli, A. Transcriptional Control of Mitochondrial Biogenesis and Function. Annu. Rev. Physiol. 2009, 71, 177–203. [Google Scholar] [CrossRef] [Green Version]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP Kinase Activity in the Regulation of PGC-1alpha Transcription in Skeletal Muscle Cells. Am. J. Physiol. Cell Physiol. 2009, 296, C116–C123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, D.; Choi, M.E. The Emerging Role of Mitophagy in Kidney Diseases. J. Life Sci. 2019, 1, 13–22. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Goh, J.-Y.; Xiao, L.; Xian, H.; Lim, K.-L.; Liou, Y.-C. Reactive Oxygen Species Trigger Parkin/PINK1 Pathway-Dependent Mitophagy by Inducing Mitochondrial Recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 Is Activated by Mitochondrial Membrane Potential Depolarization and Stimulates Parkin E3 Ligase Activity by Phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Fermín, L.M.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Tapia, E.; Rivero, I.; Pedraza-Chaverri, J. The Protective Effect of Alpha-Mangostin against Cisplatin-Induced Cell Death in LLC-PK1 Cells Is Associated to Mitochondrial Function Preservation. Antioxidants 2019, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy Is a Component of Epithelial Cell Fate in Obstructive Uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yu, S.; Zhang, K.; Zhang, Z.; Li, C.; Gao, B.; Zhang, W.; Wang, Y. Exogenous H2S Inhibits Autophagy in Unilateral Ureteral Obstruction Mouse Renal Tubule Cells by Regulating the ROS-AMPK Signaling Pathway. Cell. Physiol. Biochem. 2018, 49, 2200–2213. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
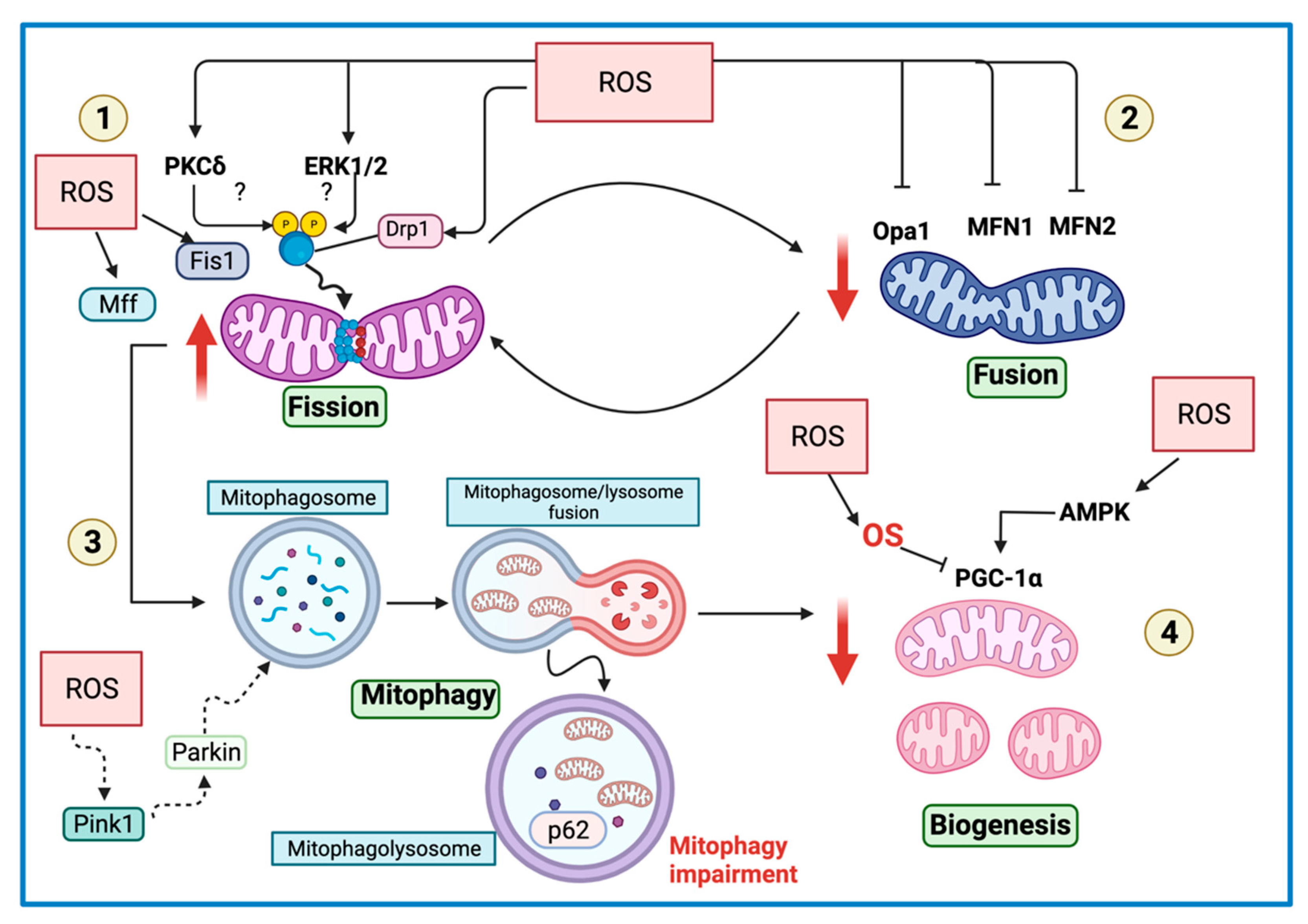{kind=link}
| AKI Model | In Vivo Model | Mitochondrial Dynamic Protein | Mechanism | References |
|---|---|---|---|---|
| Cisplatin-induced nephrotoxicity and I/R | C57BL/6 mice | ↑Drp1 | Drp1 translocates to the mitochondria in response to ROS overproduction. | Brooks et al. [163] |
| Maleate-induced nephrotoxicity | Male Wistar rats | ↑Drp1, Fis1 | Maleate-induced OS promotes mitochondrial fission by increasing Drp1 and Fis1. | Molina-Jijón et al. [84] |
| Cisplatin | C57BL/6 mice | ↓Opa1, Mfn1 ↑Fis1 ↑Pink1, parkin | ROS and mtROS promote fission and decrease the mitochondrial fusion process. | Ortega-Domínguez et al. [164] |
| I/R | C57BL/6 mice | Drp1−/− | The deletion of Drp1 improves mitochondrial function by decreasing mtROS. | Perry et al. [165] |
| Cisplatin | Female C57BL/6 Idh2−/− mice | ↓Opa1 ↑Drp1 | Idh2−/− decreases NADPH and GSH levels, inducing OS and triggering fission increase and fusion decrease. | Kong et al. [145] |
| I/R | Female C57BL/6 Idh2−/− mice | ↑Drp1, Fis1 ↓Opa1 | Idh2−/−-induced mtROS, decreasing the levels of fusion proteins and augmenting fission proteins. | Han et al. [144] |
| Folic acid | Male Wistar rats | ↑Fis1, Drp1 ↓Opa1, Mfn1 ↑Pink1, ↓LC3 ↓PGC-1α, ↓NRF1, NRF2 | ROS overproduction increases fission and reduces the fusion process. | Aparicio-Trejo et al. [44] |
| Nephrotoxicity by K2Cr2O7 | Male Wistar rats | ↑Drp1 ↓PGC-1α | ROS overproduction increases fission and reduces biogenesis. | Ávila-Rojas et al. [148] |
| MA: induced Fanconi syndrome | Male Wistar rats | ↓TFAM, ↑Fis1, Drp1 ↑Parkin, p62, LC3-II | SF prevents mitochondrial fission increase and TFAM decrease and regulates mitophagy. | Briones-Herrera et al. [109] |
| CKD Model | In Vivo Model | Mitochondrial Dynamic Proteins Alteration | Effects | References |
|---|---|---|---|---|
| DN | Male C57BL/6J mice | ↓PGC-1α, AMPK | Reduced ROS levels decrease mitochondrial biogenesis. | Dugan et al. [141] |
| 5/6 nephrectomy | Male Sprague-Dawley rats | ↓Mfn2, Opa1 ↑Drp1 ↓PPARγ | The use of pioglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) activator, decreases mtROS, improving mitochondrial dynamics. | Sun et al. [8] |
| 5/6 nephrectomy | Male Wistar rats | ↑Mfn1, Opa1 ↓Fis1, Drp1 | ROS overproduction favors mitochondrial fusion. | Aparicio-Trejo et al. [128] |
| UUO | Male C57BL/6J mice | ↑LC3, Pink1, parkin | ROS-induced senescence impairs mitophagy. | Liu et al. [169] |
| UUO | Male C57BL/6J mice | ↓Drp1 ↑LC3, Pink1, parkin | mtROS recruit Drp1 to the OMM, regulating mitophagy parkin-dependent. | Li et al. [167] |
| 5/6 Nephrectomy | Male Wistar rats | ↓NRF1, NRF2, TFAM PGC-1α, PPARα ↓Mfn2, Opa1 ↑LC3, p62 | Mitochondrial biogenesis and dynamics are altered temporal courses. | Prieto-Carrasco et al. [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. https://doi.org/10.3390/biom11081144
Aranda-Rivera AK, Cruz-Gregorio A, Aparicio-Trejo OE, Pedraza-Chaverri J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules. 2021; 11(8):1144. https://doi.org/10.3390/biom11081144
Chicago/Turabian StyleAranda-Rivera, Ana Karina, Alfredo Cruz-Gregorio, Omar Emiliano Aparicio-Trejo, and José Pedraza-Chaverri. 2021. "Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases" Biomolecules 11, no. 8: 1144. https://doi.org/10.3390/biom11081144