
Pathological Mechanism of a Constitutively Active Form of Stromal Interaction Molecule 1 in Skeletal Muscle

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
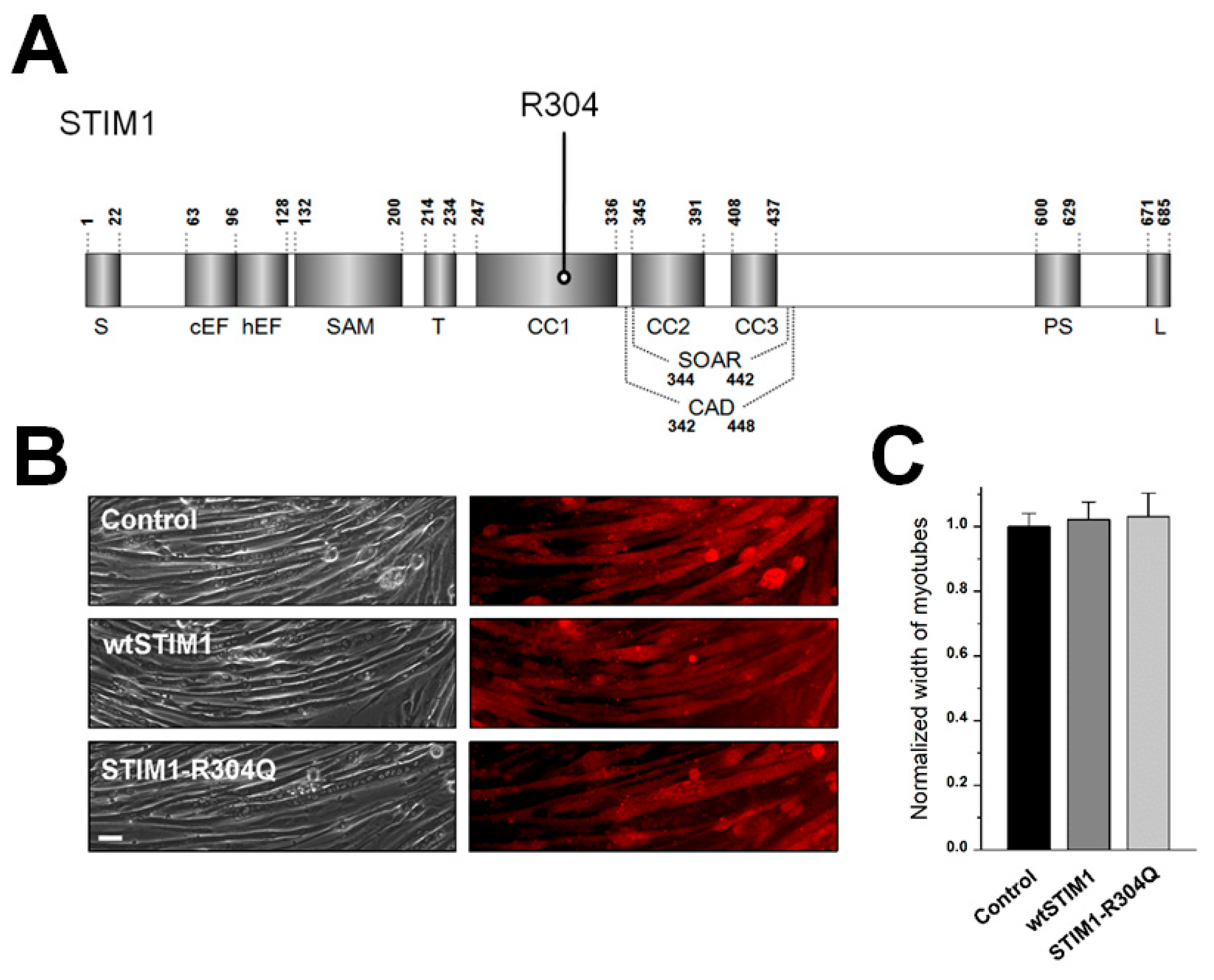
2.2. cDNA Construction, Cell Culture, and STIM1-R304Q Expression
2.3. Immunocytochemistry and Immunoblot Assays
2.4. Single-Myotube Ca2+ Imaging
2.5. Transmission Electron Microscopy (TEM) Observation, Myotube Width Measurement, and Mitochondrial Length Measurement
2.6. Statistical Analysis
3. Results and Discussion
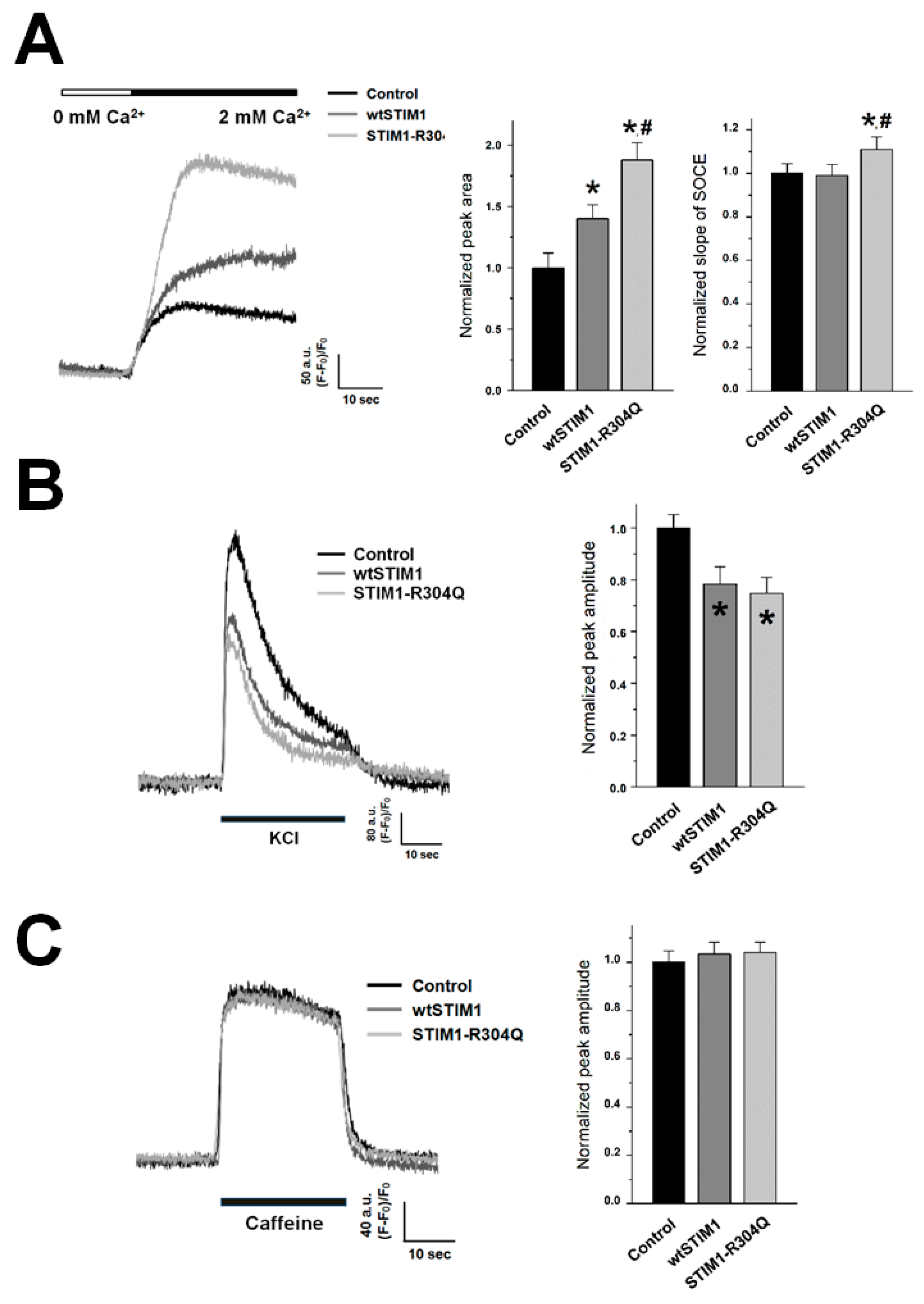
3.1. By Affecting Both the Amplitude and the Onset Rate of SOCE, STIM1-R304Q Induces Hyper-SOCE in Skeletal Myotubes
3.2. STIM1-R304Q Retains the Ability of STIM1 to Attenuate DHPR Activity in Skeletal Myotubes
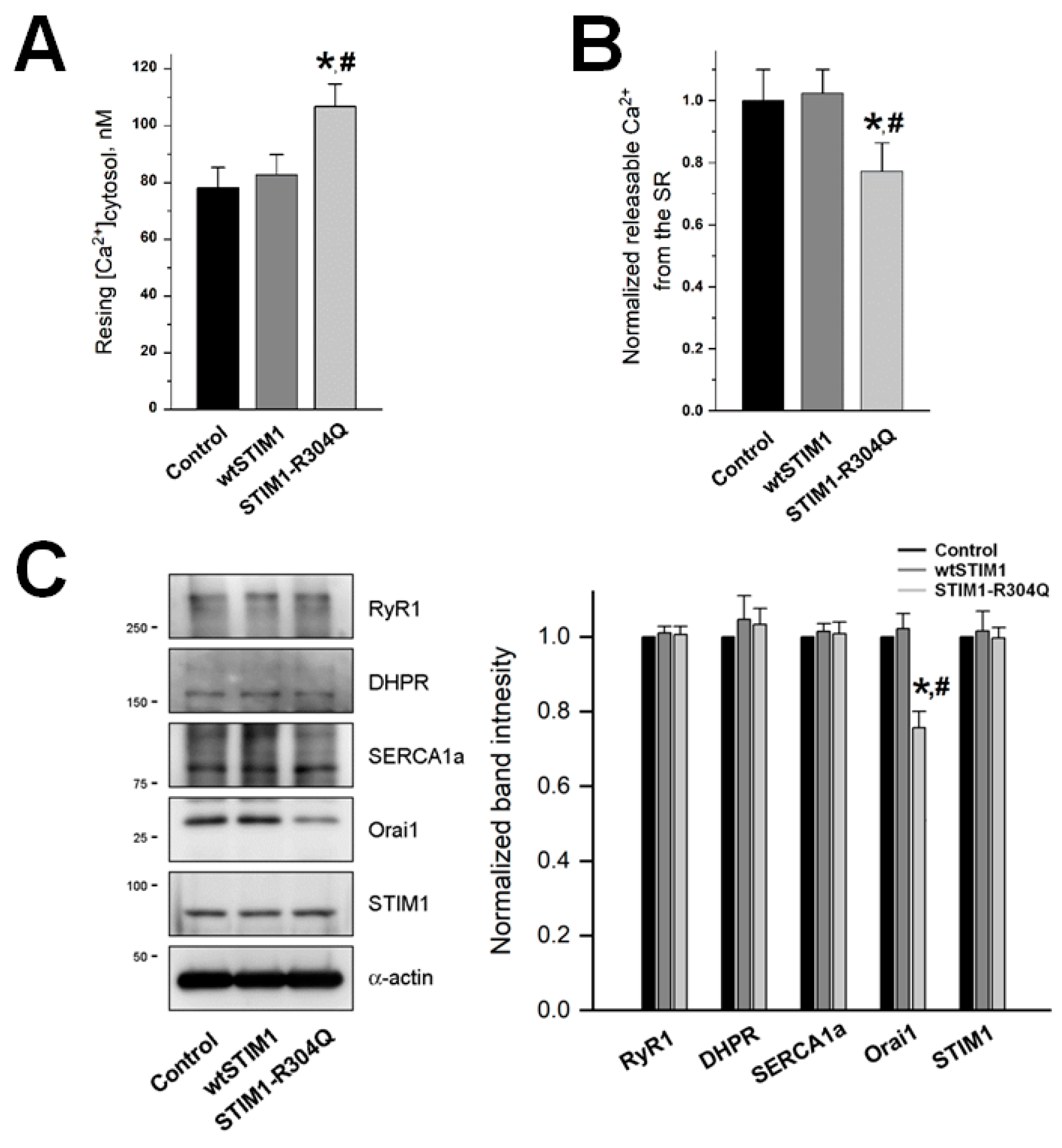
3.3. STIM1-R304Q Disturbs the Intracellular Ca2+ Distribution between the Cytosol and the SR in Skeletal Myotubes
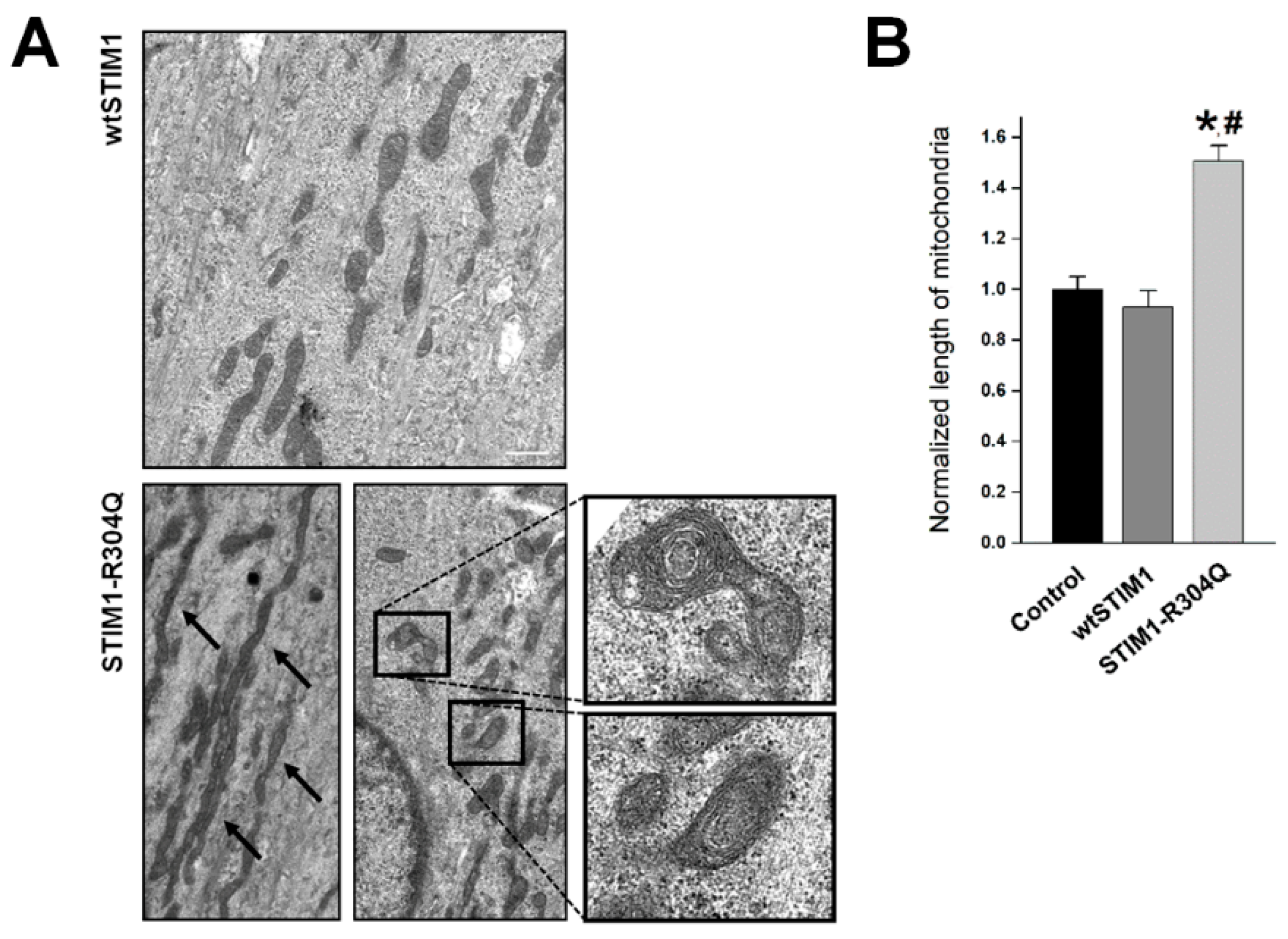
3.4. STIM1-R304Q Changes the Shape of Mitochondria in Skeletal Myotubes
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| STIM1 | Stromal interaction molecule1 |
| SOCE | Store-operated Ca2+ entry |
| EC | Excitation–contraction |
| DHPR | Dihydropyridine receptor |
| RyR1 | Ryanodine receptor 1 |
| SERCA1a | Sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1a |
References
- Lee, E.H. Ca2+ channels and skeletal muscle diseases. Prog. Biophys. Mol. Biol. 2010, 103, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Woo, J.S.; Perez, C.F.; Lee, E.H. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 2017, 49, e378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, C.H.; Lee, K.J.; Lee, E.H. With the greatest care, stromal interaction molecule (STIM) proteins verify what skeletal muscle is doing. BMB Rep. 2018, 51, 378–387. [Google Scholar] [CrossRef]
- Morin, G.; Biancalana, V.; Echaniz-Laguna, A.; Noury, J.B.; Lornage, X.; Moggio, M.; Ripolone, M.; Violano, R.; Marcorelles, P.; Marechal, D.; et al. Tubular aggregate myopathy and Stormorken syndrome: Mutation spectrum and genotype/phenotype correlation. Hum. Mutat. 2020, 41, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Stadlbauer, M.; Muik, M.; Rathner, P.; Stathopulos, P.; Ikura, M.; Muller, N.; Romanin, C. A dual mechanism promotes switching of the Stormorken STIM1 R304W mutant into the activated state. Nat. Commun. 2018, 9, 825. [Google Scholar] [CrossRef]
- Bohm, J.; Chevessier, F.; Maues De Paula, A.; Koch, C.; Attarian, S.; Feger, C.; Hantai, D.; Laforet, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Huang, M.; Hyun, C.; Oh, M.R.; Lee, K.J.; Cho, C.H.; Lee, E.H. A muscular hypotonia-associated STIM1 mutant at R429 induces abnormalities in intracellular Ca2+ movement and extracellular Ca2+ entry in skeletal muscle. Sci. Rep. 2019, 9, 19140. [Google Scholar] [CrossRef]
- Lee, K.J.; Woo, J.S.; Hwang, J.H.; Hyun, C.; Cho, C.H.; Kim, D.H.; Lee, E.H. STIM1 negatively regulates Ca2+ release from the sarcoplasmic reticulum in skeletal myotubes. Biochem. J. 2013, 453, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Borsani, O.; Piga, D.; Costa, S.; Govoni, A.; Magri, F.; Artoni, A.; Cinnante, C.M.; Fagiolari, G.; Ciscato, P.; Moggio, M.; et al. Stormorken Syndrome Caused by a p.R304W STIM1 Mutation: The First Italian Patient and a Review of the Literature. Front. Neurol. 2018, 9, 859. [Google Scholar] [CrossRef]
- Harris, E.; Burki, U.; Marini-Bettolo, C.; Neri, M.; Scotton, C.; Hudson, J.; Bertoli, M.; Evangelista, T.; Vroling, B.; Polvikoski, T.; et al. Complex phenotypes associated with STIM1 mutations in both coiled coil and EF-hand domains. Neuromuscul. Disord. 2017, 27, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Silva-Rojas, R.; Laporte, J.; Bohm, J. STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Gamage, T.H.; Gunnes, G.; Lee, R.H.; Louch, W.E.; Holmgren, A.; Bruton, J.D.; Lengle, E.; Kolstad, T.R.S.; Revold, T.; Amundsen, S.S.; et al. STIM1 R304W causes muscle degeneration and impaired platelet activation in mice. Cell Calcium 2018, 76, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A.; Blau, H.M. Methods for myoblast transplantation. Methods Cell Biol. 1997, 52, 261–272. [Google Scholar] [CrossRef]
- Lee, K.J.; Park, C.S.; Woo, J.S.; Kim, D.H.; Ma, J.; Lee, E.H. Mitsugumin 53 attenuates the activity of sarcoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) in skeletal muscle. Biochem. Biophys. Res. Commun. 2012, 428, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Cho, C.H.; Lee, K.J.; Kim, D.H.; Ma, J.; Lee, E.H. Hypertrophy in skeletal myotubes induced by junctophilin-2 mutant, Y141H, involves an increase in store-operated Ca2+ entry via Orai1. J. Biol. Chem. 2012, 287, 14336–14348. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Hyun, C.; Woo, J.S.; Park, C.S.; Kim, D.H.; Lee, E.H. Stromal interaction molecule 1 (STIM1) regulates sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) in skeletal muscle. Pflugers Arch. 2014, 466, 987–1001. [Google Scholar] [CrossRef]
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca2+ entry and intracellular Ca2+ release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.R.; Lee, K.J.; Huang, M.; Kim, J.O.; Kim, D.H.; Cho, C.H.; Lee, E.H. STIM2 regulates both intracellular Ca2+ distribution and Ca2+ movement in skeletal myotubes. Sci. Rep. 2017, 7, 17936. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Lee, K.J.; Kim, K.J.; Ahn, M.K.; Cho, C.H.; Kim, D.H.; Lee, E.H. The maintenance ability and Ca2+ availability of skeletal muscle are enhanced by sildenafil. Exp. Mol. Med. 2016, 48, e278. [Google Scholar] [CrossRef] [Green Version]
- Silva-Rojas, R.; Treves, S.; Jacobs, H.; Kessler, P.; Messaddeq, N.; Laporte, J.; Bohm, J. STIM1 over-activation generates a multi-systemic phenotype affecting the skeletal muscle, spleen, eye, skin, bones and immune system in mice. Hum. Mol. Genet. 2019, 28, 1579–1593. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef] [PubMed]
- des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Jeong, S.Y.; Oh, M.R.; Allen, P.D.; Lee, E.H. TRPCs: Influential Mediators in Skeletal Muscle. Cells 2020, 9, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neustein, H.B.; Lurie, P.R.; Dahms, B.; Takahashi, M. An X-linked recessive cardiomyopathy with abnormal mitochondria. Pediatrics 1979, 64, 24–29. [Google Scholar]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | wtSTIM1 | STIM1-R304Q | ||
|---|---|---|---|---|
| Width of myotubes | 1.00 ± 0.04 (50) | 1.02 ± 0.05 (50) | 1.03 ± 0.07 (50) | |
| SOCE | Peak area | 1.00 ± 0.12 (40) | 1.40 ± 0.11 * (40) | 1.88 ± 0.14 *, # (40) |
| Slope | 1.00 ± 0.05 (30) | 0.99 ± 0.05 (30) | 1.11 ± 0.06 *, # (30) | |
| KCl response | 1.00 ± 0.05 (70) | 0.78 ± 0.07 * (70) | 0.75 ± 0.06 * (70) | |
| Caffeine response | 1.00 ± 0.05 (70) | 1.03 ± 0.05 (70) | 1.04 ± 0.04 (70) | |
| Resting [Ca2+]cytosol, nM | 78.16 ± 7.07 (50) | 82.73 ± 7.14 (50) | 106.76 ± 7.94 *, # (50) | |
| Amount of Ca2+ releasable from the SR | 1.00 ± 0.11 (40) | 1.02 ± 0.08 (40) | 0.77 ± 0.09 *, # (40) | |
| Length of mitochondria | 1.00 ± 0.05 (62) | 0.93 ± 0.07 (61) | 1.51 ± 0.06 *, # (69) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.H.; Jeong, S.Y.; Choi, J.H.; Lee, E.H. Pathological Mechanism of a Constitutively Active Form of Stromal Interaction Molecule 1 in Skeletal Muscle. Biomolecules 2021, 11, 1064. https://doi.org/10.3390/biom11081064
Park JH, Jeong SY, Choi JH, Lee EH. Pathological Mechanism of a Constitutively Active Form of Stromal Interaction Molecule 1 in Skeletal Muscle. Biomolecules. 2021; 11(8):1064. https://doi.org/10.3390/biom11081064
Chicago/Turabian StylePark, Ji Hee, Seung Yeon Jeong, Jun Hee Choi, and Eun Hui Lee. 2021. "Pathological Mechanism of a Constitutively Active Form of Stromal Interaction Molecule 1 in Skeletal Muscle" Biomolecules 11, no. 8: 1064. https://doi.org/10.3390/biom11081064