
Pharmacokinetics and Molecular Modeling Indicate nAChRα4-Derived Peptide HAEE Goes through the Blood–Brain Barrier

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Tritium-Labeled HAEE
2.3. Administration of [3H]HAEE to Animals and Blood Sampling
2.4. HAEE Assay in Tissues Specimens
2.5. Analysis of Pharmacokinetics Data
2.6. Molecular Modeling
3. Results
3.1. In Vitro Stability Assay of Synthetic HAEE
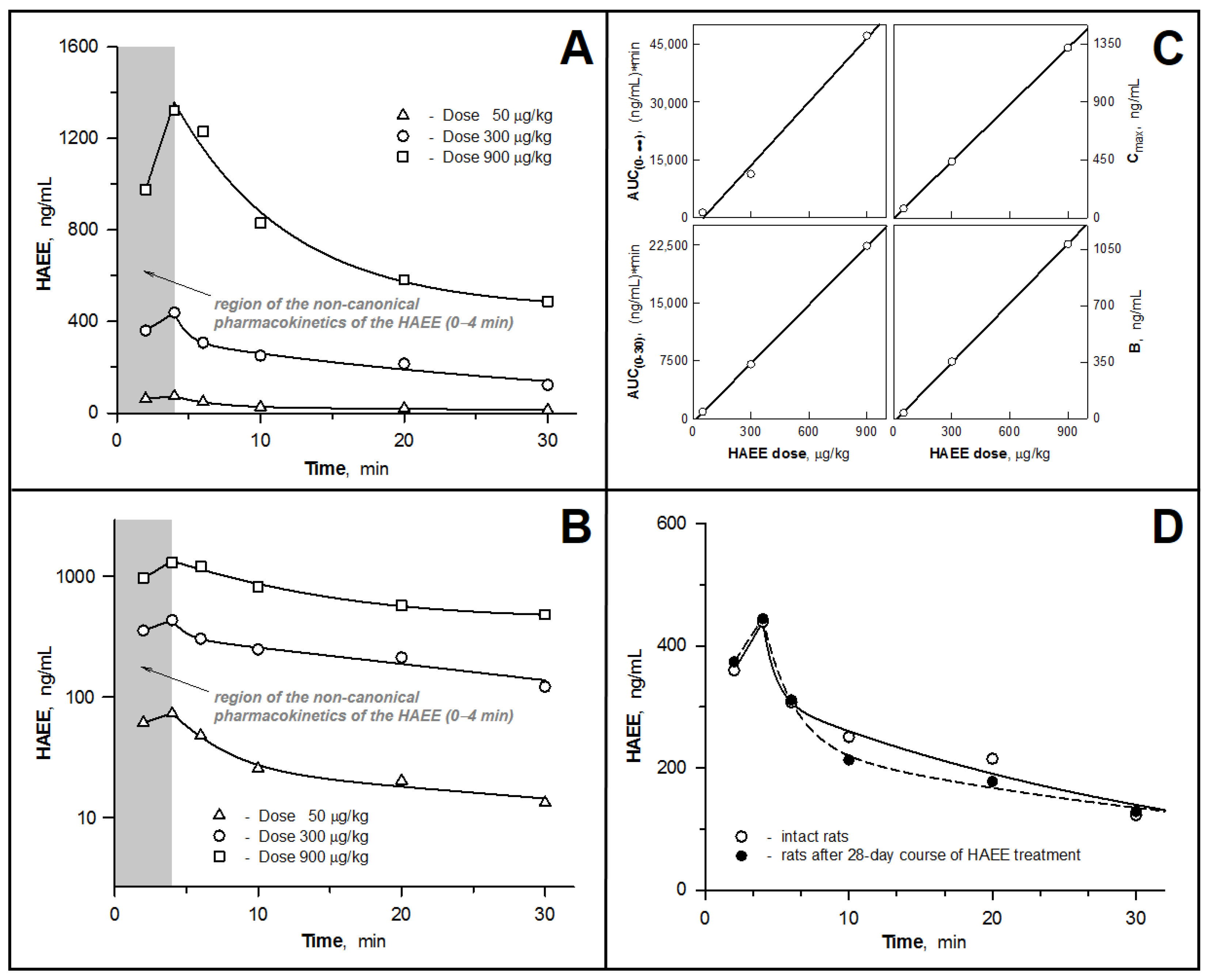
3.2. Pharmacokinetic Parameters of the HAEE Peptide after Intravenous Bolus Administration to Rabbits and Rats
3.3. Examining the Hypothesis of the HAEE Pharmacokinetics Linearity
3.4. Pharmacokinetics of HAEE after Chronic Administration of the Peptide to Rats
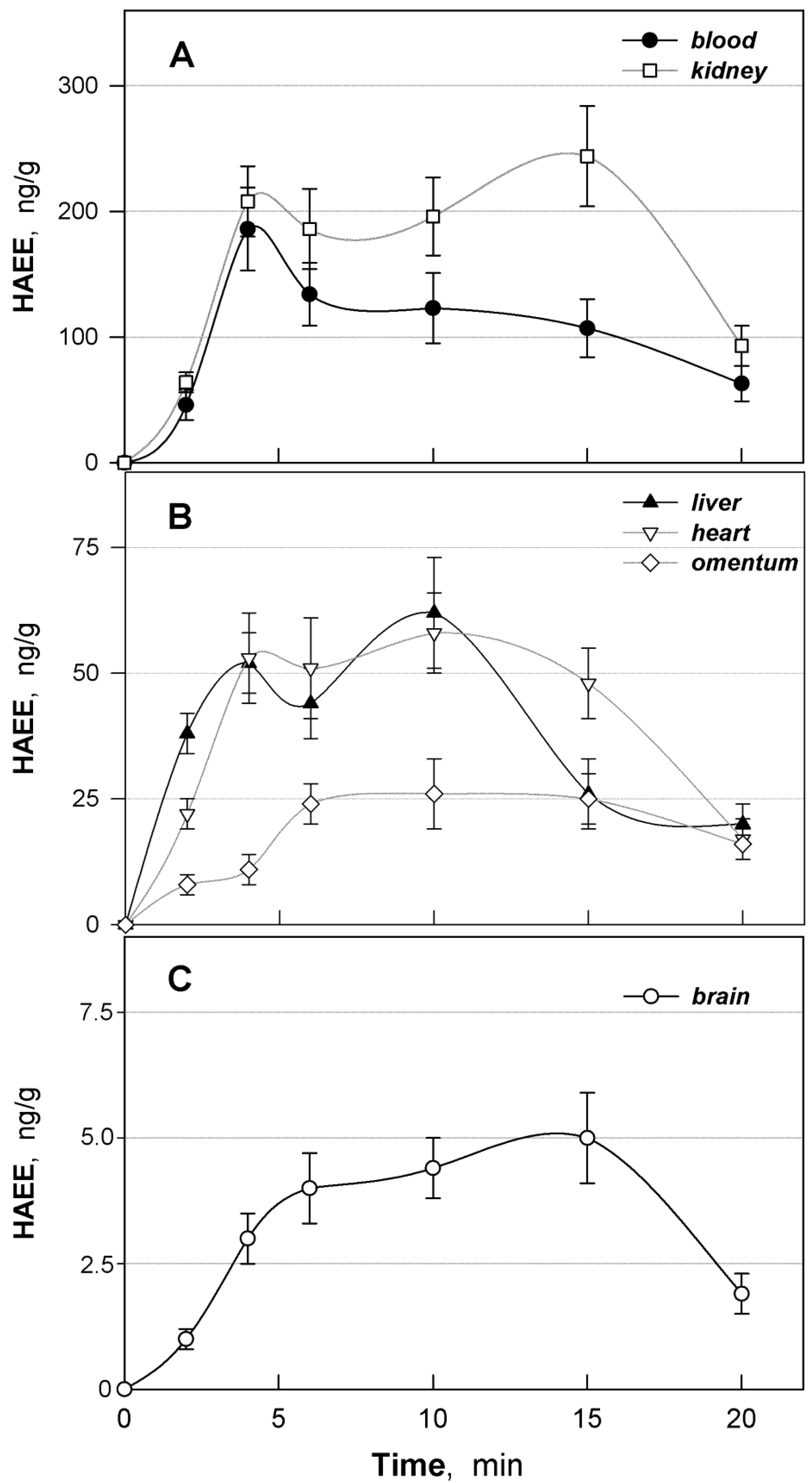
3.5. Distribution of HAEE in Mouse Tissues
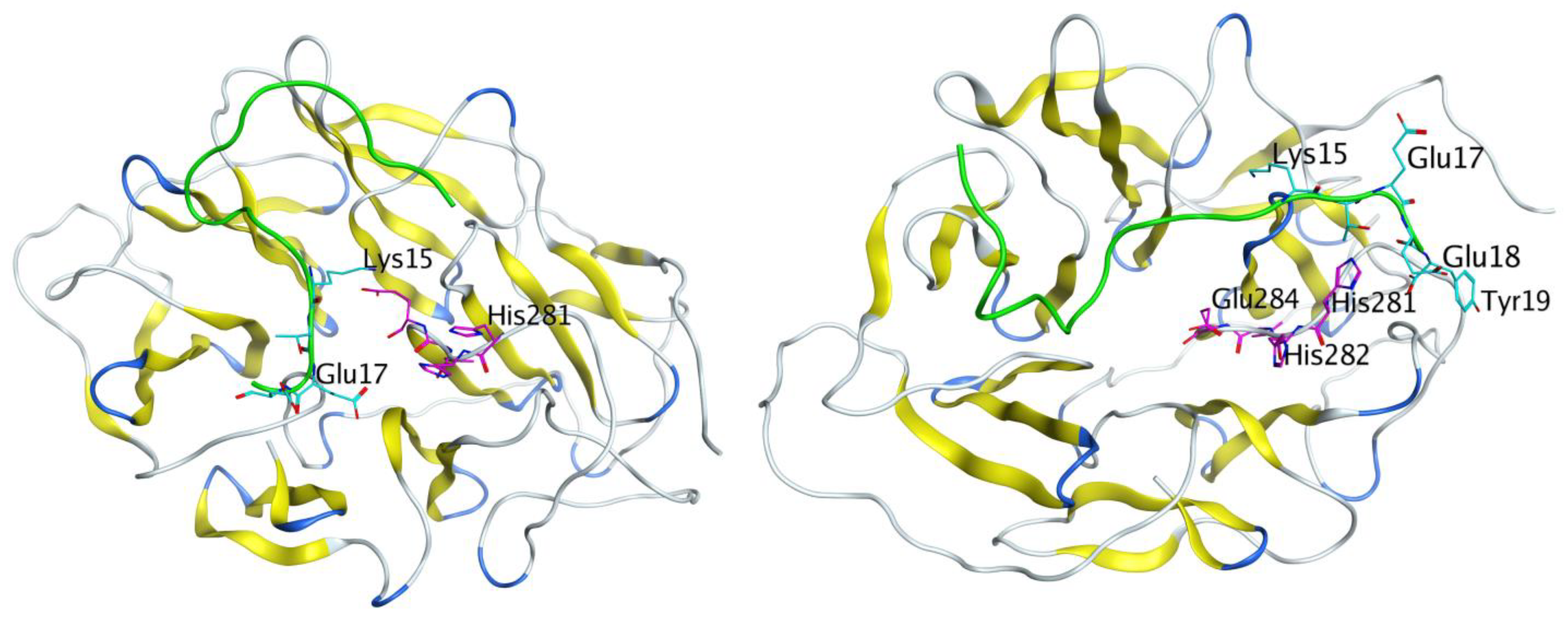
3.6. Molecular Modeling Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabri, O.; Meyer, P.M.; Gräf, S.; Hesse, S.; Wilke, S.; Becker, G.-A.; Rullmann, M.; Patt, M.; Luthardt, J.; Wagenknecht, G.; et al. Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain 2018, 141, 1840–1854. [Google Scholar] [CrossRef]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. 2020, 30, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Kozin, S.A.; Barykin, E.P.; Mitkevich, V.A.; Makarov, A.A. Anti-amyloid Therapy of Alzheimer’s Disease: Current State and Prospects. Biochemistry 2018, 83, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.L.; Tong, M.; Alfulaij, N.; Sherrin, T.; Contarino, M.; White, M.M.; Bellinger, F.P.; Todorovic, C.; Nichols, R.A. Regulation of presynaptic Ca2+, synaptic plasticity and contextual fear conditioning by a N-terminal β-amyloid fragment. J. Neurosci. 2014, 34, 14210–14218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barykin, E.; Garifulina, A.; Tolstova, A.; Anashkina, A.; Adzhubei, A.; Mezentsev, Y.; Shelukhina, I.; Kozin, S.; Tsetlin, V.; Makarov, A. Tetrapeptide Ac-HAEE-NH2 Protects α4β2 nAChR from Inhibition by Aβ. Int. J. Mol. Sci. 2020, 21, 6272. [Google Scholar] [CrossRef] [PubMed]
- Forest, K.H.; Nichols, R.A. Assessing Neuroprotective Agents for Aβ-Induced Neurotoxicity. Trends Mol. Med. 2019, 25, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Makarov, A.A. The Convergence of Alzheimer’s Disease Pathogenesis Concepts. Mol. Biol. 2019, 53, 896–903. [Google Scholar] [CrossRef]
- Tsvetkov, P.O.; Cheglakov, I.B.; Ovsepyan, A.A.; Mediannikov, O.Y.; Morozov, A.O.; Telegin, G.B.; Kozin, S.A. Peripherally Applied Synthetic Tetrapeptides HAEE and RADD Slow Down the Development of Cerebral β-Amyloidosis in AβPP/PS1 Transgenic Mice. J. Alzheimer’s Dis. 2015, 46, 849–853. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Régina, A.; Ché, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Identification and Design of Peptides as a New Drug Delivery System for the Brain. J. Pharmacol. Exp. Ther. 2007, 324, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Demeule, M.; Currie, J.-C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Zolotarev, Y.A.; Dadayan, A.K.; Borisov, Y.A.; Kozik, V.S. Solid State Isotope Exchange with Spillover Hydrogen in Organic Compounds. Chem. Rev. 2010, 110, 5425–5446. [Google Scholar] [CrossRef]
- Everett, N.B.; Simmons, B.S.; Lasher, E.P. Distribution of Blood (Fe59) and Plasma (I131) Volumes of Rats Determined by Liquid Nitrogen Freezing. Circ. Res. 1956, 4, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Zolotarev, Y.A.; Dadayan, A.K.; Kost, N.; Voevodina, M.E.; Sokolov, O.Y.; Kozik, V.S.; Shram, S.I.; Azev, V.N.; Bocharov, E.V.; Bogachouk, A.P.; et al. The qualitative analysis of the amide of the HLDF-6 peptide and its metabolites in tissues of laboratory animals with the use of tritium-labeled and deuterium-labeled derivatives. Russ. J. Bioorg. Chem. 2015, 41, 578–589. [Google Scholar] [CrossRef]
- Pieper, U.; Webb, B.M.; Dong, G.Q.; Schneidman-Duhovny, D.; Fan, H.; Kim, S.J.; Khuri, N.; Spill, Y.; Weinkam, P.; Hammel, M.; et al. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014, 42, D336–D346. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A.M. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anashkina, A.; Kravatsky, Y.; Kuznetsov, E.; Makarov, A.; Adzhubei, A. Meta-server for automatic analysis, scoring and ranking of docking models. Bioinformatics 2017, 34, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Frossard, M.-J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological Profiles of Peptide Drug Candidates for the Treatment of Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef] [Green Version]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Baig, M.H.; Ahmad, K.; Saeed, M.; Alharbi, A.M.; Barreto, G.E.; Ashraf, G.M.; Choi, I. Peptide based therapeutics and their use for the treatment of neurodegenerative and other diseases. Biomed. Pharmacother. 2018, 103, 574–581. [Google Scholar] [CrossRef]
- Ribarič, S. Peptides as Potential Therapeutics for Alzheimer’s Disease. Molecules 2018, 23, 283. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, D.A.; Hogg, D.W.; Dodsworth, T.L.; Jurado, F.R.; Read, C.C.; D’Aquila, A.L.; Barsyte-Lovejoy, D. Synthetic Peptides as Therapeutic Agents: Lessons Learned From Evolutionary Ancient Peptides and Their Transit Across Blood-Brain Barriers. Front. Endocrinol. 2019, 10, 730. [Google Scholar] [CrossRef]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to beta-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Paterson, D.; Nordberg, A. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 2000, 61, 75–111. [Google Scholar] [CrossRef]
- Wu, J.; Kuo, Y.-P.; George, A.A.; Xu, L.; Hu, J.; Lukas, R.J. β-Amyloid Directly Inhibits Human α4β2-Nicotinic Acetylcholine Receptors Heterologously Expressed in Human SH-EP1 Cells. J. Biol. Chem. 2004, 279, 37842–37851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 Binds to α7 Nicotinic Acetylcholine Receptor with High Affinity. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jürgensen, S.; Ferreira, S.T. Nicotinic Receptors, Amyloid-β, and Synaptic Failure in Alzheimer’s Disease. J. Mol. Neurosci. 2009, 40, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Parri, H.; Dineley, K.; Parri, R. Nicotinic Acetylcholine Receptor Interaction with β -Amyloid: Molecular, Cellular, and Physiological Consequences. Curr. Alzheimer Res. 2010, 7, 27–39. [Google Scholar] [CrossRef]
- Kelly, T.D. Beta-amyloid peptide—Nicotinic acetylcholine receptor interaction: The two faces of health and disease. Front. Biosci. 2007, 12, 5030–5038. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parri, H.R.; Hernandez, C.M.; Dineley, K.T. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 2011, 82, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.L.; Stokoe, S.A.; Roberts, J.P.; Sathler, M.F.; Nip, K.A.; Shou, J.; Ko, K.; Tsunoda, S.; Kim, S. Co-activation of selective nicotinic acetylcholine receptors is required to reverse beta amyloid–induced Ca2+ hyperexcitation. Neurobiol. Aging 2019, 84, 166–177. [Google Scholar] [CrossRef]
- Forest, K.H.; Alfulaij, N.; Arora, K.; Taketa, R.; Sherrin, T.; Todorovic, C.; Lawrence, J.L.M.; Yoshikawa, G.T.; Ng, H.; Hruby, V.J.; et al. Protection against β-amyloid neurotoxicity by a non-toxic endogenous N-terminal β-amyloid fragment and its active hexapeptide core sequence. J. Neurochem. 2018, 144, 201–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.P.; Stokoe, S.A.; Sathler, M.F.; Nichols, R.A.; Kim, S. Selective coactivation of α7- and α4β2-nicotinic acetylcholine receptors reverses beta-amyloid–induced synaptic dysfunction. J. Biol. Chem. 2021, 296, 100402. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Nuttall, S.D.; Robert, R.; Caine, J.M.; Dolezal, O.; Hattarki, M.; Pearce, L.A.; Davydova, N.; Masters, C.L.; Varghese, J.N.; et al. Structural studies of the tethered N-terminus of the Alzheimer’s disease amyloid-β peptide. Proteins Struct. Funct. Bioinform. 2013, 81, 1748–1758. [Google Scholar] [CrossRef]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Ségalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural Changes of Region 1-16 of the Alzheimer Disease Amyloid β-Peptide upon Zinc Binding and in Vitro Aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [Green Version]
- Istrate, A.N.; Kozin, S.A.; Zhokhov, S.S.; Mantsyzov, A.B.; Kechko, O.I.; Pastore, A.; Makarov, A.A.; Polshakov, V. Interplay of histidine residues of the Alzheimer’s disease Aβ peptide governs its Zn-induced oligomerization. Sci. Rep. 2016, 6, 21734. [Google Scholar] [CrossRef] [Green Version]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, A.W.; Sychterz, C.; Ho, M.Y.; Weber, A.; Valko, K.; Negash, K. Interrogating the relationship between rat in vivo tissue distribution and drug property data for >200 structurally unrelated molecules. Pharmacol. Res. Perspect. 2015, 3, e00173. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Pardridge, W.M. Alzheimer’s disease: Future drug development and the blood-brain barrier. Expert Opin. Investig. Drugs 2019, 28, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.P.; Torrado, M.; Custódio, B.; Silva-Reis, S.C.; Santos, S.D.; Leiro, V.; Pêgo, A.P. Breaking Barriers: Bioinspired Strategies for Targeted Neuronal Delivery to the Central Nervous System. Pharmaceutics 2020, 12, 192. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Group | Animal Species | n | Study Type | Dose of HAEE, μg/kg | HAEE Route of Administration |
|---|---|---|---|---|---|
| 1 | Rabbit | 3 | Pharmacokinetics in blood | 120 | i.v. |
| 2 | Rat | 6 | Pharmacokinetics in blood (dose dependence) | 50 | i.v. |
| 3 | Rat | 6 | 300 | i.v. | |
| 4 | Rat | 6 | 900 | i.v. | |
| 5 | Rat | 6 | Pharmacokinetics in blood (effect of chronic administration a) | 300 | i.v. |
| 6 | Mouse | 36 | Tissue distribution | 300 | i.p. |
| PK Parameter a | Units | Dose of Peptide, μg/kg | ||||
| Rabbit | Rat | |||||
| 120 | 50 | 300 | 300 b | 900 | ||
| D | ng | 440,000 | 18,000 | 105,000 | 105,000 | 315,000 |
| Cmax | ng/mL | 255 | 73.9 | 439 | 444 | 1322 |
| Tmax | min | 4 | 4 | 4 | 4 | 4 |
| AUC(0–∞) | (ng/mL) × min | 4421 | 1383 | 11,340 | 12,137 | 47,133 |
| ClT | mL/min | 100 | 13.0 | 9.3 | 8.7 | 6.7 |
| MRT | min | 29 | 33 | 32 | 42 | 36 |
| Kel | min−1 | 0.035 | 0.031 | 0.031 | 0.024 | 0.028 |
| T1/2(el) | min | 20 | 23 | 22 | 29 | 25 |
| Vd(c) | mL | 862 | 107 | 122 | 111 | 108 |
| Vd(β) | mL | 2860 | 425 | 299 | 362 | 237 |
| Vd(extrap) | mL | 3194 | 504 | 296 | 382 | 291 |
| Organ/Tissue | a AUC(0–20), (ng/g) × min | fT | fT × 100% |
|---|---|---|---|
| Blood | 2110 | 1.000 | 100.0 |
| Kidney | 3437 | 1.628 | 162.8 |
| Liver | 777 | 0.368 | 36.8 |
| Heart | 845 | 0.400 | 40.0 |
| Omentum | 395 | 0.187 | 18.7 |
| Brain | 70 | 0.033 | 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotarev, Y.A.; Mitkevich, V.A.; Shram, S.I.; Adzhubei, A.A.; Tolstova, A.P.; Talibov, O.B.; Dadayan, A.K.; Myasoyedov, N.F.; Makarov, A.A.; Kozin, S.A. Pharmacokinetics and Molecular Modeling Indicate nAChRα4-Derived Peptide HAEE Goes through the Blood–Brain Barrier. Biomolecules 2021, 11, 909. https://doi.org/10.3390/biom11060909
Zolotarev YA, Mitkevich VA, Shram SI, Adzhubei AA, Tolstova AP, Talibov OB, Dadayan AK, Myasoyedov NF, Makarov AA, Kozin SA. Pharmacokinetics and Molecular Modeling Indicate nAChRα4-Derived Peptide HAEE Goes through the Blood–Brain Barrier. Biomolecules. 2021; 11(6):909. https://doi.org/10.3390/biom11060909
Chicago/Turabian StyleZolotarev, Yurii A., Vladimir A. Mitkevich, Stanislav I. Shram, Alexei A. Adzhubei, Anna P. Tolstova, Oleg B. Talibov, Alexander K. Dadayan, Nikolai F. Myasoyedov, Alexander A. Makarov, and Sergey A. Kozin. 2021. "Pharmacokinetics and Molecular Modeling Indicate nAChRα4-Derived Peptide HAEE Goes through the Blood–Brain Barrier" Biomolecules 11, no. 6: 909. https://doi.org/10.3390/biom11060909