
Relationship between Oxidative Stress and Imatinib Resistance in Model Chronic Myeloid Leukemia Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells
2.3. MTT Assay for Cell Viability
2.4. Detection of BCR-ABL1 Presence
2.5. Detection of BCR-ABL Y253H Mutation
2.6. Assessment of ROS Level
2.7. Assessment of DNA Damage and Repair
2.8. Cell Extracts
2.9. Assessment of GSH/GSSG Levels
2.10. Activity of Glutathione Peroxidase
2.11. Activity of Catalase
2.12. Mitochondrial Membrane Potential
2.13. Statistical Analyses
3. Results
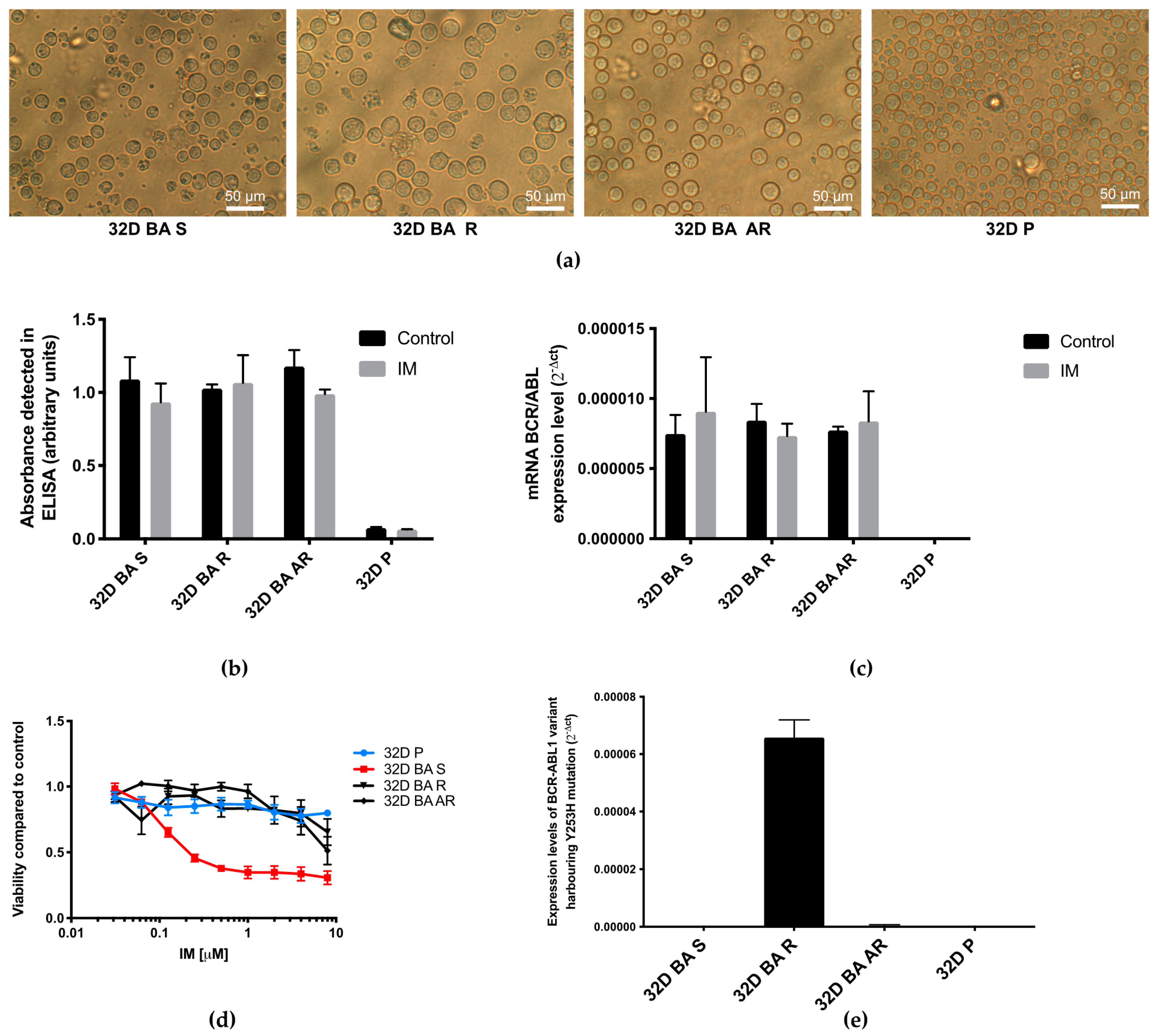
3.1. Detection of BCR-ABL1 Presence and Its Mutational Status
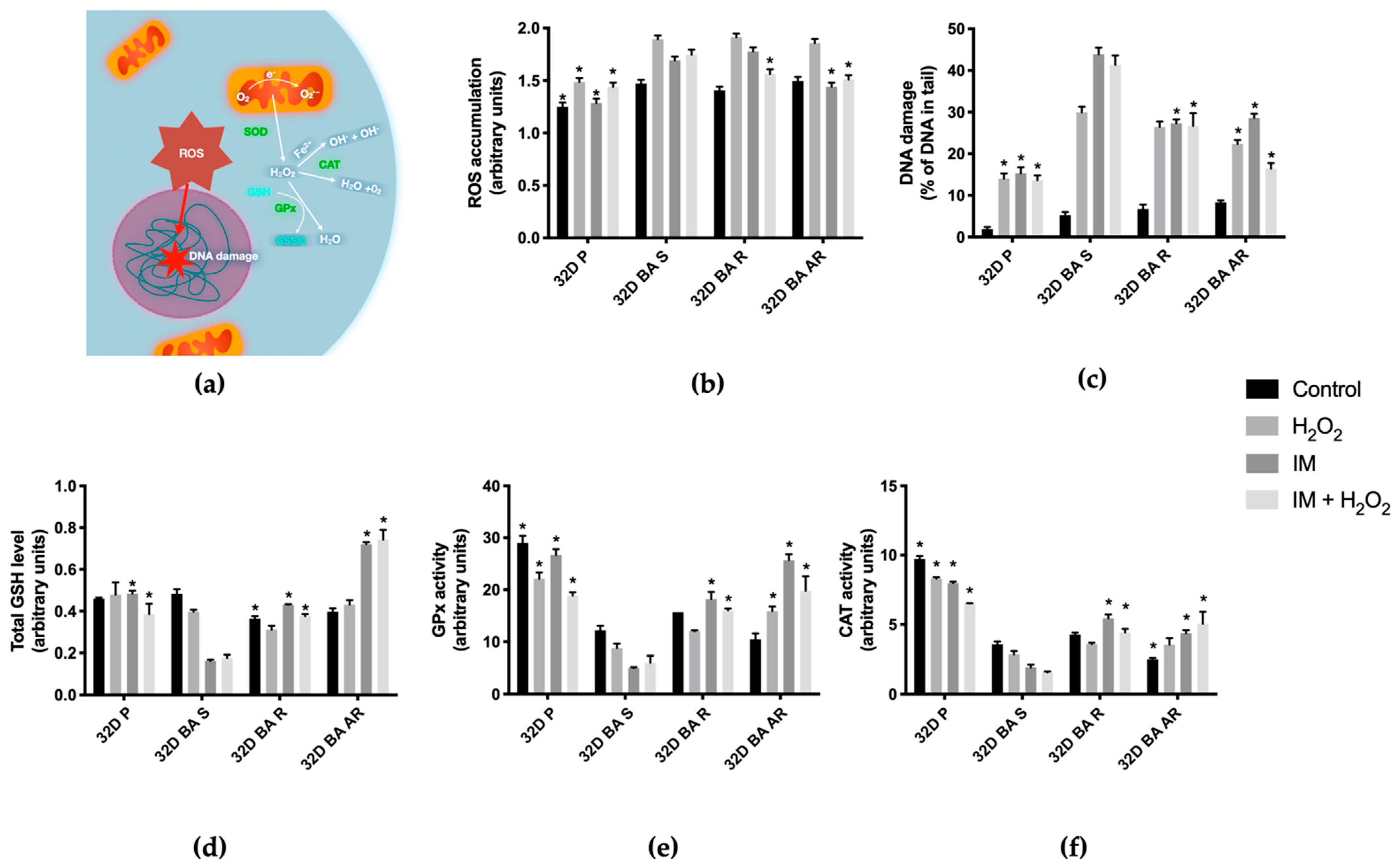
3.2. Reactive Oxygen Species Levels Are Increased in BCR-ABL1 Positive Cells Susceptible to IM When Compared to BCR-ABL1 Negative Cells, and Are Lower in Cells with IM-Acquired Resistance after Treatment with IM
3.3. Exposure to IM and/or H2O2 Increases DNA Damage in IM-Susceptible Cells
3.4. GSH Level Decreases in IM-Susceptible Cells after Exposure to IM When Compared to IM-Resistant Cells
3.5. Enzymes Involved in ROS Metabolism Are More Active in IM-resistant Cells after Exposure to IM
3.5.1. Glutathione Peroxidase
3.5.2. Catalase
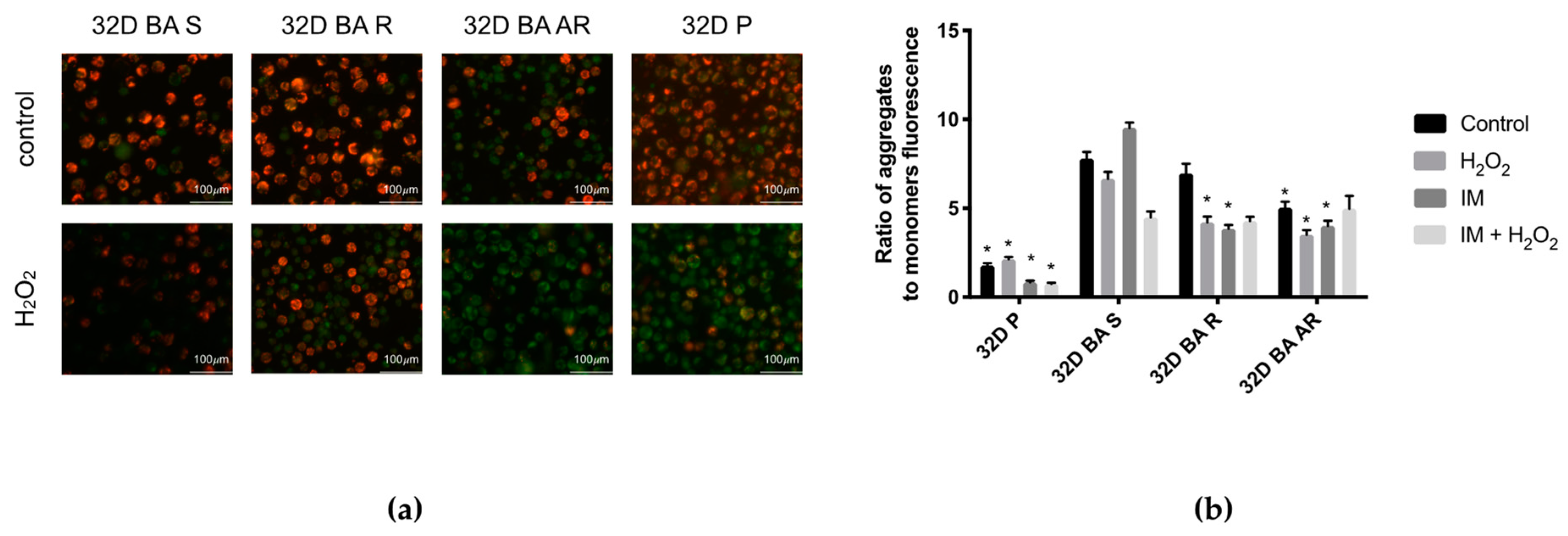
3.6. Mitochondrial Membrane Potential Is Lower in IM-Resistant Cells When Compared to IM-Susceptible Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef] [Green Version]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer Res. 2018, 138, 71–98. [Google Scholar]
- Trela, E.; Glowacki, S.; Błasiak, J. Therapy of Chronic Myeloid Leukemia: Twilight of the Imatinib Era? ISRN Oncol. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Stella, S.; Tirrò, E.; Romano, C.; Pennisi, M.S.; Puma, A.; Manzella, L.; Zanghì, A.; Stagno, F.; Di Raimondo, F.; et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol. Cancer 2018, 17, 56. [Google Scholar] [CrossRef] [Green Version]
- Talati, C.; Pinilla-Ibarz, J. Resistance in chronic myeloid leukemia: Definitions and novel therapeutic agents. Curr. Opin. Hematol. 2018, 25, 154–161. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Gao, T.; Lei, Q.; Zhang, L.; Yao, Y.; Xiong, J. Piperlongumine Induces Apoptosis in Human Melanoma Cells Via Reactive Oxygen Species Mediated Mitochondria Disruption. Nutr. Cancer 2018, 70, 502–511. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Sahoo, S.K. Exploitation of redox discrepancy in leukemia cells by a reactive oxygen species nanoscavenger for inducing cytotoxicity in imatinib resistant cells. J. Colloid Interface Sci. 2016, 467, 180–191. [Google Scholar] [CrossRef]
- Synowiec, E.; Hoser, G.; Bialkowska-Warzecha, J.; Pawlowska, E.; Skorski, T.; Blasiak, J. Doxorubicin Differentially Induces Apoptosis, Expression of Mitochondrial Apoptosis-Related Genes, and Mitochondrial Potential in BCR-ABL1-Expressing Cells Sensitive and Resistant to Imatinib. Biomed. Res. Int. 2015, 2015, 673512–673519. [Google Scholar] [CrossRef] [Green Version]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.-D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.M.; Fernandes, M.S.; Salgia, R.; Levine, R.L.; Griffin, J.D.; Sattler, M. NADPH oxidases regulate cell growth and migration in myeloid cells transformed by oncogenic tyrosine kinases. Leukemia 2011, 25, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.M.; Irwin, M.E.; Gao, Y.; Ban, K.; Shi, P.; Arlinghaus, R.B.; Amin, H.M.; Chandra, J. Inhibition of the NADPH oxidase regulates heme oxygenase 1 expression in chronic myeloid leukemia. Cancer 2012, 118, 3433–3445. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.-T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH oxidase-generated hydrogen peroxide induces DNA damage in mutant FLT3-expressing leukemia cells. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef] [Green Version]
- Kruk, J.; Aboul-Enein, H.Y. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef]
- Azad, N.; Iyer, A.; Vallyathan, V.; Wang, L.; Castranova, V.; Stehlik, C.; Rojanasakul, Y. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann. N. Y. Acad. Sci. 2010, 1203, 1–6. [Google Scholar] [CrossRef]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Antoszewska-Smith, J.; Pawlowska, E.; Blasiak, J. Reactive oxygen species in BCR-ABL1-expressing cells-relevance to chronic myeloid leukemia. Acta Biochim. Pol. 2017, 64, 1–10. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Wasik, M.A.; Slupianek, A.; Salomoni, P.; Kitamura, T.; Calabretta, B.; Skorski, T. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 1999, 189, 1229–1242. [Google Scholar] [CrossRef]
- Slupianek, A.; Hoser, G.; Majsterek, I.; Bronisz, A.; Malecki, M.; Blasiak, J.; Fishel, R.; Skorski, T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol. Cell. Biol. 2002, 22, 4189–4201. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Halprin, K.M.; Ohkawara, A. The measurement of glutathione in human epidermis using glutathione reductase. J. Investig. Dermatol. 1967, 48, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Synowiec, E.; Hoser, G.; Wojcik, K.; Pawlowska, E.; Skorski, T.; Błasiak, J. UV Differentially Induces Oxidative Stress, DNA Damage and Apoptosis in BCR-ABL1-Positive Cells Sensitive and Resistant to Imatinib. Int. J. Mol. Sci. 2015, 16, 18111–18128. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Giannoudis, A.; Lane, S.; Williamson, P.; Pirmohamed, M.; Clark, R.E. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin. Pharmacol. Ther. 2008, 83, 258–264. [Google Scholar] [CrossRef]
- Pascu, E.G.; Gaman, M.A.; Moisa, C.; Assani, A.D.; Gaman, A.M. The involvement of oxidative stress in Chronic Myeloid Leukemia. Rom. Biotechnol. Lett. 2020, 25, 1267–1274. [Google Scholar] [CrossRef]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Kwee, J.K.; Luque, D.G.; Ferreira, A.C.D.S.; Vasconcelos, F.D.C.; Silva, K.L.; Klumb, C.E.; Maia, R.C. Modulation of reactive oxygen species by antioxidants in chronic myeloid leukemia cells enhances imatinib sensitivity through survivin downregulation. Anticancer Drugs 2008, 19, 975–981. [Google Scholar] [CrossRef]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef]
- Blasiak, J.; Hoser, G.; Bialkowska-Warzecha, J.; Pawlowska, E.; Skorski, T. Reactive Oxygen Species and Mitochondrial DNA Damage and Repair in BCR-ABL1 Cells Resistant to Imatinib. BioRes. Open Access 2015, 4, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Pascu, E.G.; Gaman, M.A.; Moisa, C.; Gaman, A.M. Oxidative stress and BCR-ABL1 transcript levels in chronic myeloid leukemia: An intricate relationship. Rev. Chim. 2019, 70, 3193–3196. [Google Scholar] [CrossRef]
- Ko, B.W.; Han, J.; Heo, J.Y.; Jang, Y.; Kim, S.J.; Kim, J.; Lee, M.J.; Ryu, M.J.; Song, I.C.; Jo, Y.S.; et al. Metabolic characterization of imatinib-resistant BCR-ABL T315I chronic myeloid leukemia cells indicates down-regulation of glycolytic pathway and low ROS production. Leuk. Lymphoma 2016, 57, 2180–2188. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Maulik, N.; Das, D.K. Redox signaling in vascular angiogenesis. Free Radic. Biol. Med. 2002, 33, 1047–1060. [Google Scholar] [CrossRef]
- Aslan, M.; Ozben, T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid. Redox Signal. 2003, 5, 781–788. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.H.; Chang, H.-J.; Cho, J.Y.; Chun, H.S. Cytoprotective effect of anthocyanins against doxorubicin-induced toxicity in H9c2 cardiomyocytes in relation to their antioxidant activities. Food Chem. Toxicol. 2007, 45, 1873–1881. [Google Scholar] [CrossRef]
- Singh, S.; Khan, A.R.; Gupta, A.K. Role of glutathione in cancer pathophysiology and therapeutic interventions. J. Exp. Ther. Oncol. 2012, 9, 303–316. [Google Scholar]
- Yang, P.; Ebbert, J.O.; Sun, Z.; Weinshilboum, R.M. Role of the glutathione metabolic pathway in lung cancer treatment and prognosis: A review. J. Clin. Oncol. 2006, 24, 1761–1769. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Habermann, K.J.; Grünewald, L.; van Wijk, S.; Fulda, S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell Death Dis. 2017, 8, e3067. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Zhang, G.-N.; Barbuti, A.M.; Zhang, X.; Karadkhelkar, N.; Zhou, J.; Ding, K.; Pan, J.; Yoganathan, S.; Yang, D.-H.; et al. Preclinical development of a novel BCR-ABL T315I inhibitor against chronic myeloid leukemia. Cancer Lett. 2020, 472, 132–141. [Google Scholar] [CrossRef]
- Yao, D.-M.; Zhou, J.-D.; Zhang, Y.-Y.; Yang, L.; Wen, X.-M.; Yang, J.; Guo, H.; Chen, Q.; Lin, J.; Qian, J. GPX3 promoter is methylated in chronic myeloid leukemia. Int. J. Clin. Exp. Pathol. 2015, 8, 6450–6457. [Google Scholar] [PubMed]
- Kos, Z.; Pavelić, L.; Pekić, B.; Pavelić, K. Reversal of human myeloid leukemia cells into normal granulocytes and macrophages: Activity and intracellular distribution of catalase. Oncology 1987, 44, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Juul, T.; Malolepszy, A.; Dybkaer, K.; Kidmose, R.; Rasmussen, J.T.; Andersen, G.R.; Johnsen, H.E.; Jørgensen, J.-E.; Andersen, S.U. The in vivo toxicity of hydroxyurea depends on its direct target catalase. J. Biol. Chem. 2010, 285, 21411–21415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrax, J.; Pedrosa, R.C.; Beck, R.; Dejeans, N.; Taper, H.; Calderon, P.B. In situ modulation of oxidative stress: A novel and efficient strategy to kill cancer cells. Curr. Med. Chem. 2009, 16, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Pedrosa, R.C.; Dejeans, N.; Glorieux, C.; Levêque, P.; Gallez, B.; Taper, H.; Eeckhoudt, S.; Knoops, L.; Calderon, P.B.; et al. Ascorbate/menadione-induced oxidative stress kills cancer cells that express normal or mutated forms of the oncogenic protein Bcr-Abl. An in vitro and in vivo mechanistic study. Investig. New Drugs 2011, 29, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Song, L.-L.; Tu, Y.-Y.; Xia, L.; Wang, W.-W.; Wei, W.; Ma, C.-M.; Wen, D.-H.; Lei, H.; Xu, H.-Z.; Wu, Y.-L. Targeting catalase but not peroxiredoxins enhances arsenic trioxide-induced apoptosis in K562 cells. PLoS ONE 2014, 9, e104985. [Google Scholar] [CrossRef] [Green Version]
- Coe, E.; Schimmer, A.D. Catalase activity and arsenic sensitivity in acute leukemia. Leuk. Lymphoma 2008, 49, 1976–1981. [Google Scholar] [CrossRef]
- Mutlu Altundağ, E.; Yılmaz, A.M.; Koçtürk, S.; Taga, Y.; Yalçın, A.S. Synergistic Induction of Apoptosis by Quercetin and Curcumin in Chronic Myeloid Leukemia (K562) Cells. Nutr. Cancer 2018, 70, 97–108. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Q.; Zhang, L.; Zhong, Y.; Fan, G.; Zhang, Z.; Wang, R.; Jin, M.; Qiu, Y.; Kong, D. Stellettin B induces apoptosis in human chronic myeloid leukemia cells via targeting PI3K and Stat5. Oncotarget 2017, 8, 28906–28921. [Google Scholar] [CrossRef] [Green Version]
- Giannoudis, A.; Giannoudis, A.; Davies, A.; Davies, A.; Lucas, C.M.; Lucas, C.M.; Harris, R.J.; Harris, R.J.; Pirmohamed, M.; Pirmohamed, M.; et al. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): Implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood 2008, 112, 3348–3354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active transport of imatinib into and out of cells: Implications for drug resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S. Mitochondria: A target for cancer therapy. Br. J. Pharmacol. 2006, 147, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545. [Google Scholar] [CrossRef]
- Gier, B.; Krippeit-Drews, P.; Sheiko, T.; Aguilar-Bryan, L.; Bryan, J.; Düfer, M.; Drews, G. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J. Clin. Investig. 2009, 119, 3246–3256. [Google Scholar]
- Chatterjee, A.; Dasgupta, S.; Sidransky, D. Mitochondrial subversion in cancer. Cancer Prev. Res. 2011, 4, 638–654. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Głowacki, S.; Synowiec, E.; Szwed, M.; Toma, M.; Skorski, T.; Śliwiński, T. Relationship between Oxidative Stress and Imatinib Resistance in Model Chronic Myeloid Leukemia Cells. Biomolecules 2021, 11, 610. https://doi.org/10.3390/biom11040610
Głowacki S, Synowiec E, Szwed M, Toma M, Skorski T, Śliwiński T. Relationship between Oxidative Stress and Imatinib Resistance in Model Chronic Myeloid Leukemia Cells. Biomolecules. 2021; 11(4):610. https://doi.org/10.3390/biom11040610
Chicago/Turabian StyleGłowacki, Sylwester, Ewelina Synowiec, Marzena Szwed, Monika Toma, Tomasz Skorski, and Tomasz Śliwiński. 2021. "Relationship between Oxidative Stress and Imatinib Resistance in Model Chronic Myeloid Leukemia Cells" Biomolecules 11, no. 4: 610. https://doi.org/10.3390/biom11040610