
Assessing Various Control Samples for Microarray Gene Expression Profiling of Laryngeal Squamous Cell Carcinoma

 , , , ,
, , , ,
Abstract
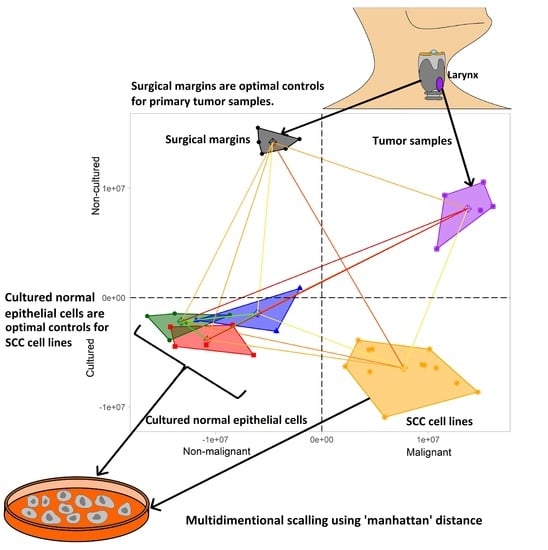
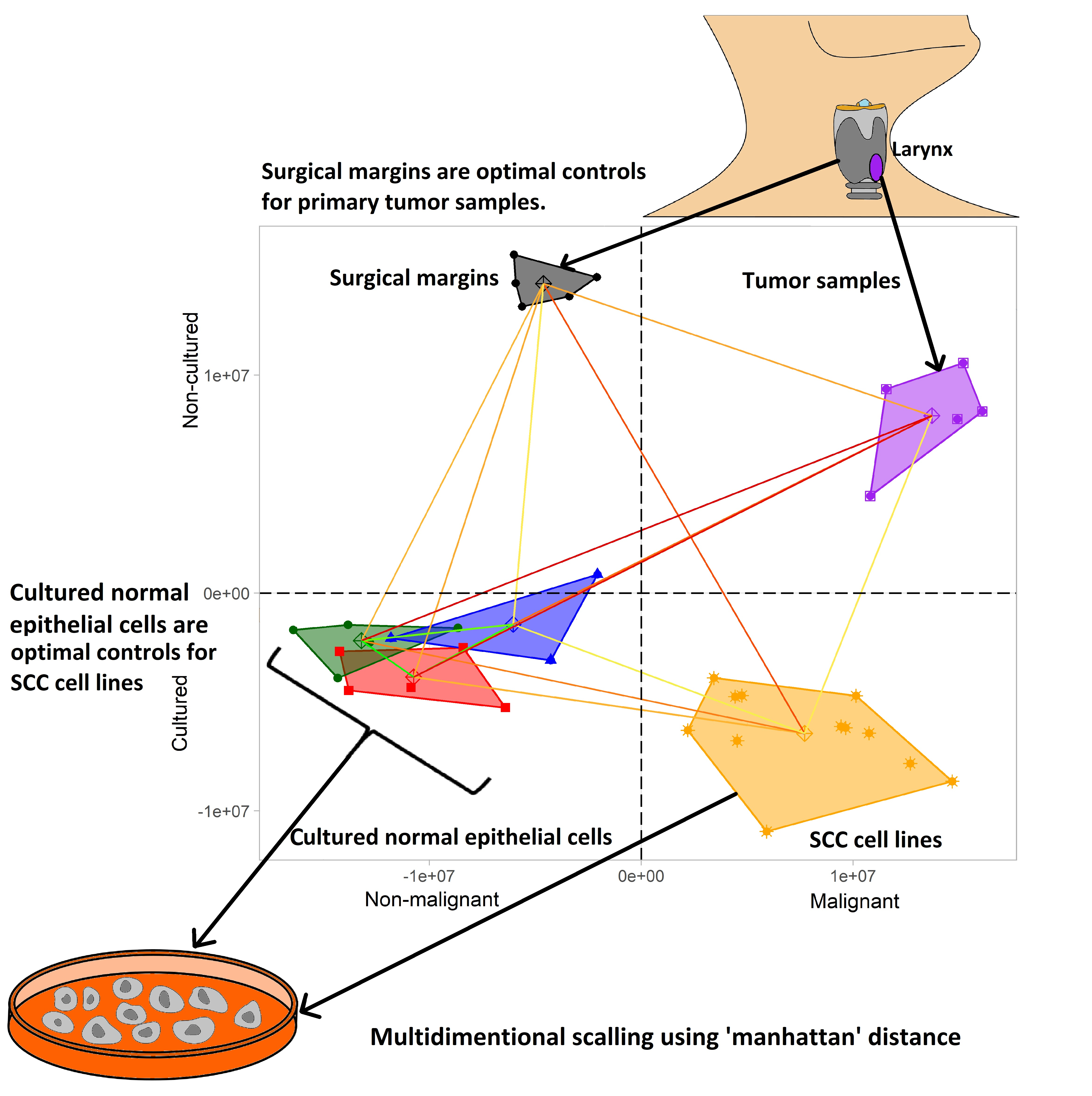
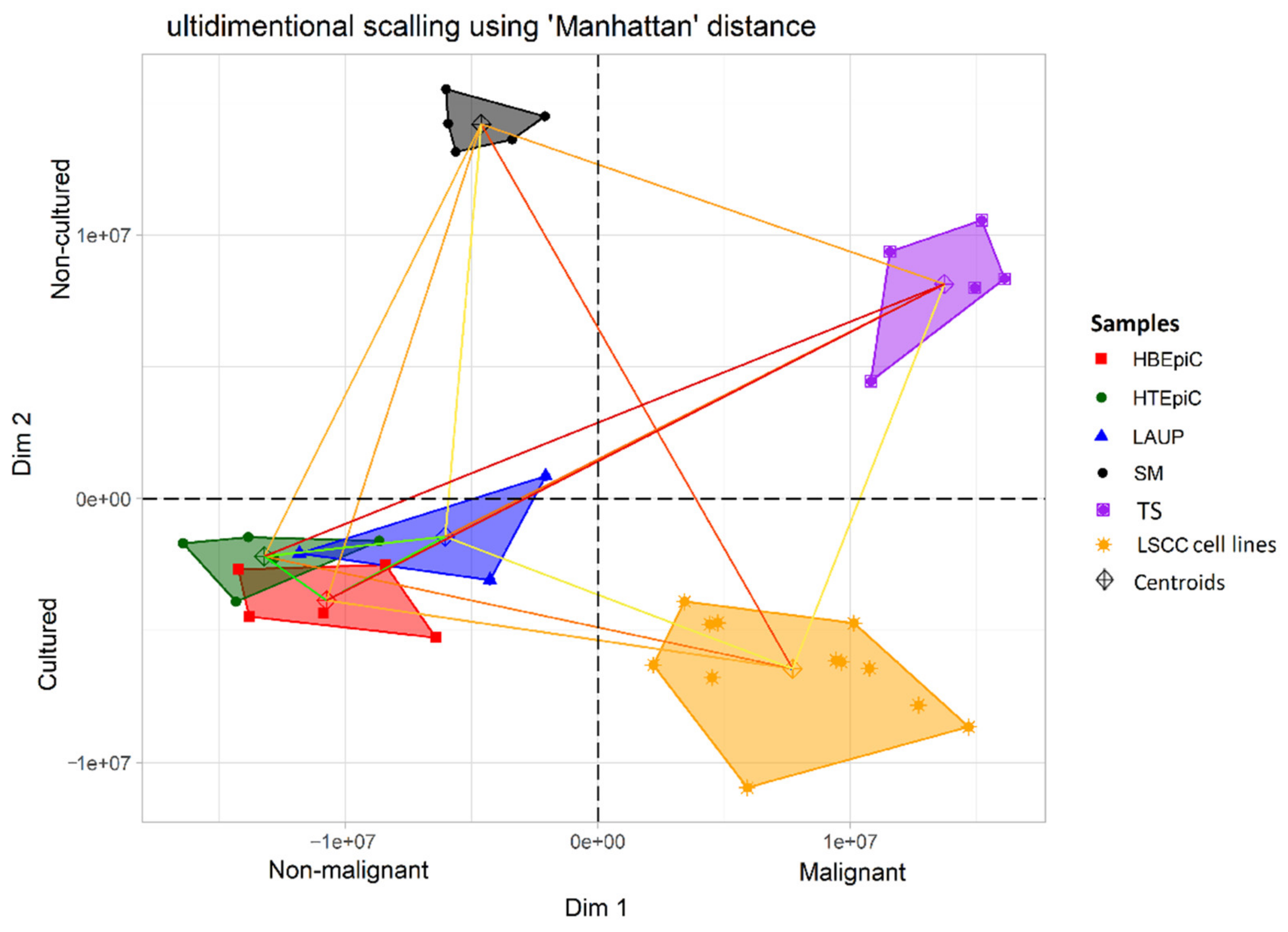
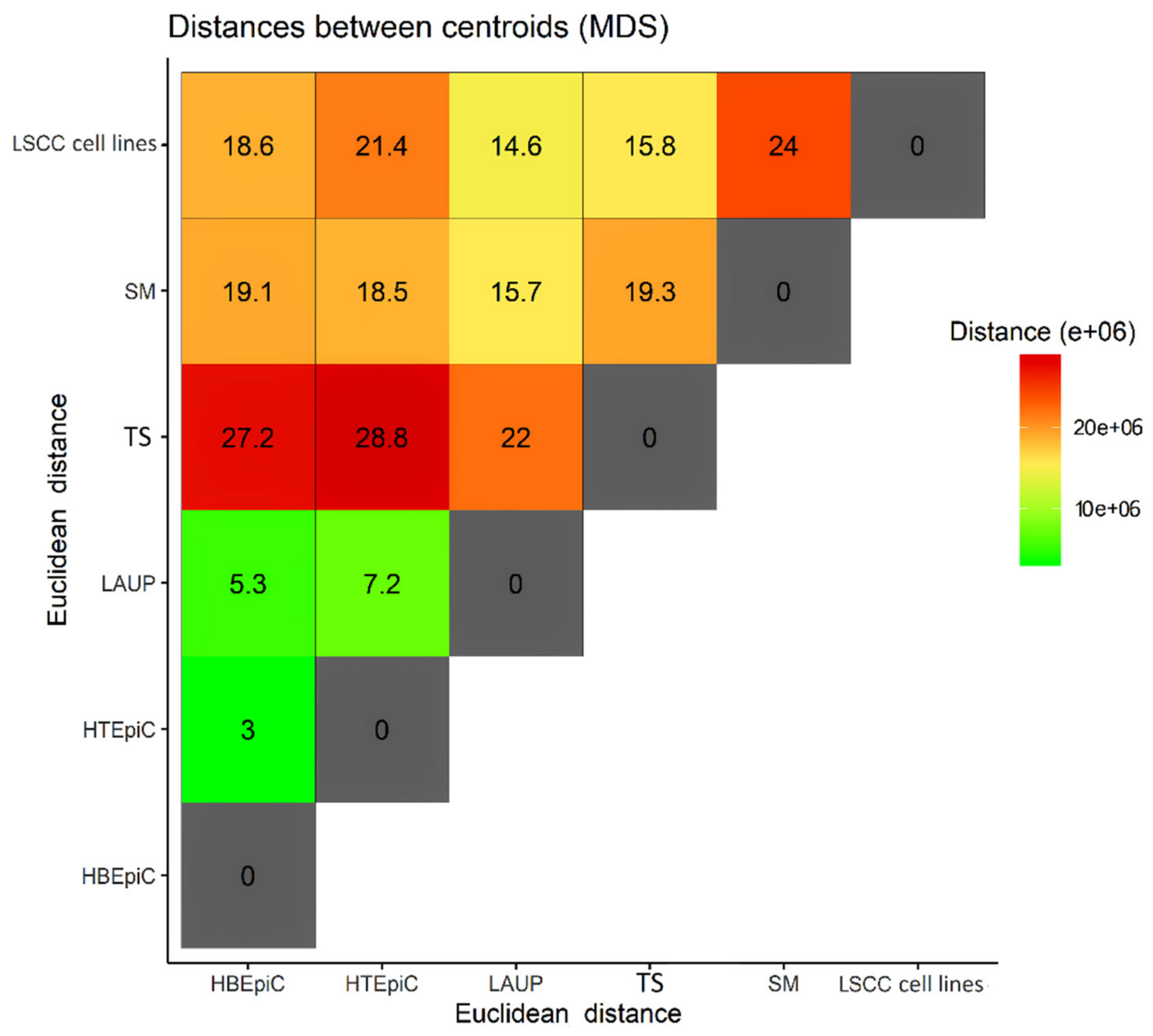
:
1. Introduction
2. Materials and Methods
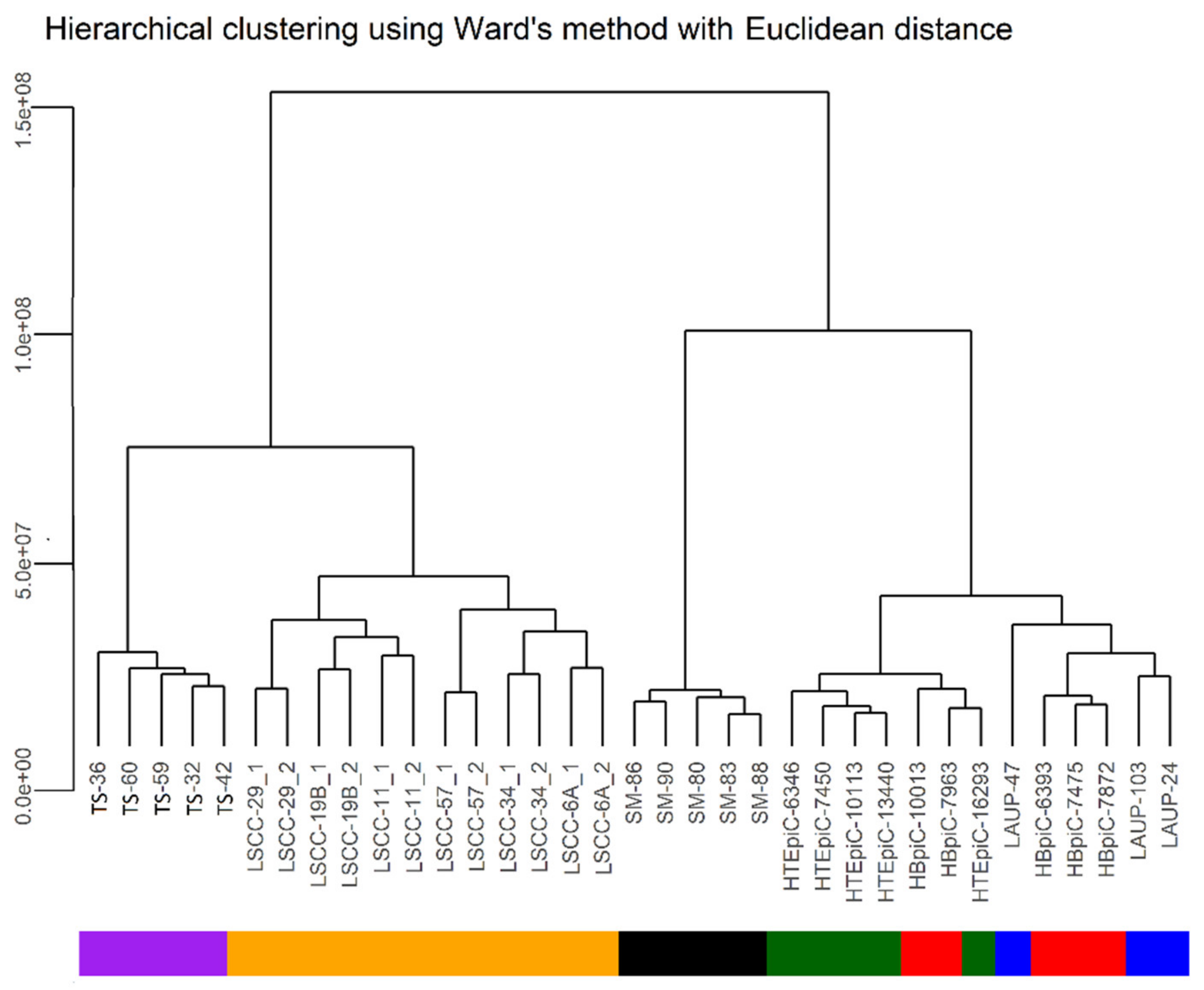
2.1. Control Samples Used for Gene Expression Profiling
- Human bronchial epithelial cells (HBEpiC, n = 5) and human tracheal epithelial cells (HTEpiC, n = 5), (frozen, cryopreserved cells at passage 0/1; ScienCell Research Laboratories, Carlsbad, CA, USA). The cells were cultured according to the protocol provided by ScienCell using the recommended Bronchial Epithelial Cell Medium (ScienCell Research Laboratories, Carlsbad, CA, USA) with 10% fetal bovine serum in T75 and T25 FLASK (Sarstedt Inc, Nümbrecht, Germany) coated with poly-L-lysine (PLL, ScienCell Research Laboratories, Carlsbad, CA, USA). The second passage, at confluence of approximately 70%, was used for expression profiling.
- Normal squamous cells were obtained from noncancerous patients using laser assisted uvulopalatoplasty (LAUP, n = 3). Cells were cultured prior to RNA isolation (approved by Ethical Review Board, no. 1156/18) in 25 cm2 flasks coated with 0.3 mg/mL PureCol collagen (Nutacon, Leimuiden, The Netherlands) in Dulbecco’s Modified Eagle Medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 20% fetal bovine serum (Biochrom, Polgen, Łodz, Poland). The first passage of proliferating cells was used for expression profiling.
- Tumor surgical margins (n = 5) obtained during laryngectomy of LSCC patients treated at the Department of Otolaryngology at the University of Medical Sciences in Poznan, Poland. The specimens were fresh frozen and tissues lacking cancer cells according to histological examination were analyzed. The Institutional Ethical Review of the University of Medical Sciences approved tissue collection (no. 904/06), and informed consent was obtained from the patients.
2.2. Tumor Samples Used for Gene Expression Profiling
- LSCC tumor samples (n = 5) obtained during laryngectomy from patients treated at the Department of Otolaryngology University of Medical Sciences in Poznan, Poland. The specimens were characterized by pathological examination. Fresh frozen samples were used for RNA isolation. The Institutional Ethical Review of the University of Medical Sciences approved tissue collection (no. 904/06), and informed consent was obtained from the patients.
- Cultured in vitro six LSCC cell lines (n = 6) (UT-SCC-6A, UT-SCC-11, UT-SCC-19B, UT-SCC-29, UT-SCC-34 and UT-SCC-57 [12,13,14,15]) established at the University of Turku (Finland). The cells were grown in 25 cm2 flasks in Dulbecco’s Modified Eagle Medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Biochrom, Polgen, Lodz, Poland). The gene expression profile of each LSCC cell line was performed twice (two separate microarray experiments) in order to obtain biological replicates.
2.3. RNA Isolation and Microarray Analysis
2.4. Bioinformatics
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, R.; Ochs, M.F.; Ahn, S.M.; Hennessey, P.; Tan, M.; Soudry, E.; Gaykalova, D.A.; Uemura, M.; Brait, M.; Shao, C.; et al. Expression Microarray Analysis Reveals Alternative Splicing of LAMA3 and DST Genes in Head and Neck Squamous Cell Carcinoma. PLoS ONE 2014, 9, e91263. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Michailidi, C.; Marchionni, L.; Pickering, C.R.; Frederick, M.J.; Myers, J.N.; Yegnasubramanian, S.; Hadar, T.; Noordhuis, M.G.; Zizkova, V.; et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics 2014, 9, 1031–1046. [Google Scholar] [CrossRef] [Green Version]
- Korampalli, T.S.; Green, V.L.; Greenman, J.; Stafford, N.D. Protein profiling of angiogenesis-related growth factors in laryngeal carcinoma: Pattern of protein expression in relation to tumour progression. Int. J. Oncol. 2011, 39, 1033–1039. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Roy, S.; Kar, M.; Padhi, S.; Saha, A.; Anuja, K.; Banerjee, B. Role of p38 MAPK in disease relapse and therapeutic resistance by maintenance of cancer stem cells in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Beibei, Y.; Rong, Y.; Yunfei, Y.; Wenchao, Z. Research Progress Regarding Surgical Margins, Molecular Margins, and Prognosis of Laryngeal Carcinoma. Ear Nose Throat J. 2020, 145561320903146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strzelczyk, J.K.; Krakowczyk, L.; Gołąbek, K.; Owczarek, A.J. Expression profiles of selected genes in tumors and matched surgical margins in oral cavity cancer: Do we have to pay attention to the molecular analysis of the surgical margins? Adv. Clin. Exp. Med. 2018, 27, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Baart, V.M.; Van Duijn, C.; Van Egmond, S.L.; Dijckmeester, W.A.; Jansen, J.C.; Vahrmeijer, A.L.; Sier, C.F.M.; Cohen, D. EGFR and αvβ6 as Promising Targets for Molecular Imaging of Cutaneous and Mucosal Squamous Cell Carcinoma of the Head and Neck Region. Cancers 2020, 12, 1474. [Google Scholar] [CrossRef]
- Dickson, M.A.; Hahn, W.C.; Ino, Y.; Ronfard, V.; Wu, J.Y.; Weinberg, R.A.; Louis, D.N.; Li, F.P.; Rheinwald, J.G. Human Keratinocytes That Express hTERT and Also Bypass a p16INK4a-Enforced Mechanism That Limits Life Span Become Immortal yet Retain Normal Growth and Differentiation Characteristics. Mol. Cell. Biol. 2000, 20, 1436–1447. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, J.; Szaumkessel, M.; Kostrzewska-Poczekaj, M.; Bednarek, K.; Paczkowska, J.; Jackowska, J.; Grenman, R.; Szyfter, K.; Wierzbicka, M.; Giefing, M.; et al. Global miRNA Expression Profiling Identifies miR-1290 as Novel Potential oncomiR in Laryngeal Carcinoma. PLoS ONE 2015, 10, e0144924. [Google Scholar] [CrossRef]
- Hout, M.C.; Papesh, M.H.; Goldinger, S.D. Multidimensional scaling. Wiley Interdiscip. Rev. Cogn. Sci. 2013, 4, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Guess, M.J.; Wilson, S.B. Introduction to hierarchical clustering. J. Clin. Neurophysiol. 2002, 19, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Servomaa, K.; Kiuru, A.; Grenman, R.; Pekkola-Heino, K.; Pulkkinen, J.O.; Rytomaa, T. p53 mutations associated with increased sensitivity to ionizing radiation in human head and neck cancer cell lines. Cell Prolif. 1996, 29, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Järvinen, A.K.; Autio, R.; Haapa-Paananen, S.; Wolf, M.; Saarela, M.; Grénman, R.; Leivo, I.; Kallioniemi, O.; Mäkitie, A.A.; Monni, O. Identification of target genes in laryngeal squamous cell carcinoma by high-resolution copy number and gene expression microarray analyses. Oncogene 2006, 25, 6997–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarmuz, M.; Golusinski, W.; Grenman, R.; Szyfter, K. Analysis of chromosome aberrations in cell lines derived from laryngeal cancer in relation to tumor progression. Eur. Arch. Oto-Rhino-Laryngol. 2002, 259, 269–273. [Google Scholar] [CrossRef]
- Jarmuz, M.; Grenman, R.; Golusinski, W.; Szyfter, K. Aberrations of 11q13 in laryngeal squamous cell lines and their prognostic significance. Cancer Genet. Cytogenet. 2005, 160, 82–88. [Google Scholar] [CrossRef]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532-4–536-7. [Google Scholar]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 1 January 2019).
- Wickham, H. Reshaping Data with thereshapePackage. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Mao, L.; Clark, D. Molecular margin of surgical resections-Where do we go from here? Cancer 2015, 121, 1914–1916. [Google Scholar] [CrossRef]
- Koffler, J.; Sharma, S.; Hess, J. Predictive value of epigenetic alterations in head and neck squamous cell carcinoma. Mol. Cell. Oncol. 2014, 1, e954827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Biological Process Name | Accession Number | Gene Ontology Resource (FDR) | DAVID (FDR) | Differentially Expressed Genes Involved in the Process (Common for DAVID and Gene Ontology Resource Tools) |
|---|---|---|---|---|
| Angiogenesis | GO:0001525 | 4.08 × 10−14 | 5.20 × 10−13 | ACVRL1, ANGPT1, ANGPT2, ANPEP, APOD, CALCRL, CCL2, CEACAM1, COL15A1, COL18A1, COL8A2, CXCL17, ECM1, ECSCR, ENPEP, EPHB2, ERAP1, FGF10, FGF18, FLT1, HEY1, HMOX1, HOXA3, HOXA7, HOXB3, JAM3, KDR, LEP, LEPR, MEOX2, MMP19, MMP2, NDNF, NOV, NRXN1, NRXN3, PDE3B, PIK3CG, PLXDC1, PLXND1, PRKD1, PTEN, PTPRB, RAMP2, ROBO4, RORA, S1PR1, SERPINE1, SHC1, SOX17, SOX18, TAL1, TEK, THSD7A, THY1, TIE1, TMEM100, TNFRSF12A, TNFSF12, VASH1, VAV3 |
| Immune response | GO:0006955 | 5.36 × 10−50 | 3.99 × 10−58 | ACKR4, ADAMDEC1, AIM2, C1QC, C1R, C3, C5AR1, C7, CCL13, CCL14, CCL18, CCL19, CCL2, CCL21, CCL22, CCL23, CCL26, CCL3, CCL4, CCL5, CCL8, CCR1, CCR2, CCR5, CCR6, CCR7, CD1A, CD1E, CD27, CD36, CD40LG, CD7, CD74, CD79B, CD86, CD8B, CD96, CFP, CLNK, CMKLR1, CRIP1, CST7, CTLA4, CTSG, CTSS, CTSW, CX3CL1, CXCL12, CXCL13, CXCL14, CXCL9, EDA, ENDOU, ENPP1, ENPP2, FCGR1A, FCGR1B, FCGR2B, FCGR2C, FCGR3A, FCGR3B, FYB, GBP2, GBP6, GEM, GPR183, GPR65, GZMA, HLA-B, HLA-C, HLA-DMA, HLA-DMB, HLA-DOA, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DQB2, HLA-DRA, HLA-DRB1, HLA-DRB3, HLA-DRB4, HLA-DRB5, IGHA1, IGHA2, IGHD, IGHV1-69, IGHV3-13, IGHV3-23, IGHV3-30, IGHV3-33, IGHV3-48, IGHV3-53, IGKC, IGKV1-17, IGKV1-39, IGKV1-5, IGKV1D-39, IGKV2D-28, IGKV4-1, IGLC1, IGLV1-44, IGLV2-14, IGLV3-1, IGLV3-19, IGLV3-21, IGLV3-25, IGSF6, IL10, IL15, IL16, IL1A, IL1B, IL1R1, IL1R2, IL2RA, IL2RG, IL32, IL36A, IL7, IL7R, IRF8, JCHAIN, LAT, LCP2, LILRB2, LST1, LTB, LY75, MARCH1, MBP, MS4A2, NRROS, OAS1, OAS2, PKHD1L1, RGS1, SAMHD1, SEMA3C, SEMA4D, SMAD6, TENM1, TGFBR3, THBS1, TLR1, TLR10, TLR4, TNFRSF1B, TNFSF11, TNFSF12, TNFSF13B, TNFSF8, TRBC1, TRGC2, WAS, ZAP70 |
| Response to wounding | GO:0009611 | 6.86 × 10−13 | 5.06 × 10−3 | CCR1, CCR2, CX3CR1, DST, F2RL2, FGF7, GAP43, GRIN2A, HHEX, ITGB4, KLK6, LYVE1, PLLP, SLC1A2, SOX2, VASH1, VWF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustaszewski, A.; Kostrzewska-Poczekaj, M.; Janiszewska, J.; Jarmuz-Szymczak, M.; Wierzbicka, M.; Marszal, J.; Grénman, R.; Giefing, M. Assessing Various Control Samples for Microarray Gene Expression Profiling of Laryngeal Squamous Cell Carcinoma. Biomolecules 2021, 11, 588. https://doi.org/10.3390/biom11040588
Ustaszewski A, Kostrzewska-Poczekaj M, Janiszewska J, Jarmuz-Szymczak M, Wierzbicka M, Marszal J, Grénman R, Giefing M. Assessing Various Control Samples for Microarray Gene Expression Profiling of Laryngeal Squamous Cell Carcinoma. Biomolecules. 2021; 11(4):588. https://doi.org/10.3390/biom11040588
Chicago/Turabian StyleUstaszewski, Adam, Magdalena Kostrzewska-Poczekaj, Joanna Janiszewska, Malgorzata Jarmuz-Szymczak, Malgorzata Wierzbicka, Joanna Marszal, Reidar Grénman, and Maciej Giefing. 2021. "Assessing Various Control Samples for Microarray Gene Expression Profiling of Laryngeal Squamous Cell Carcinoma" Biomolecules 11, no. 4: 588. https://doi.org/10.3390/biom11040588