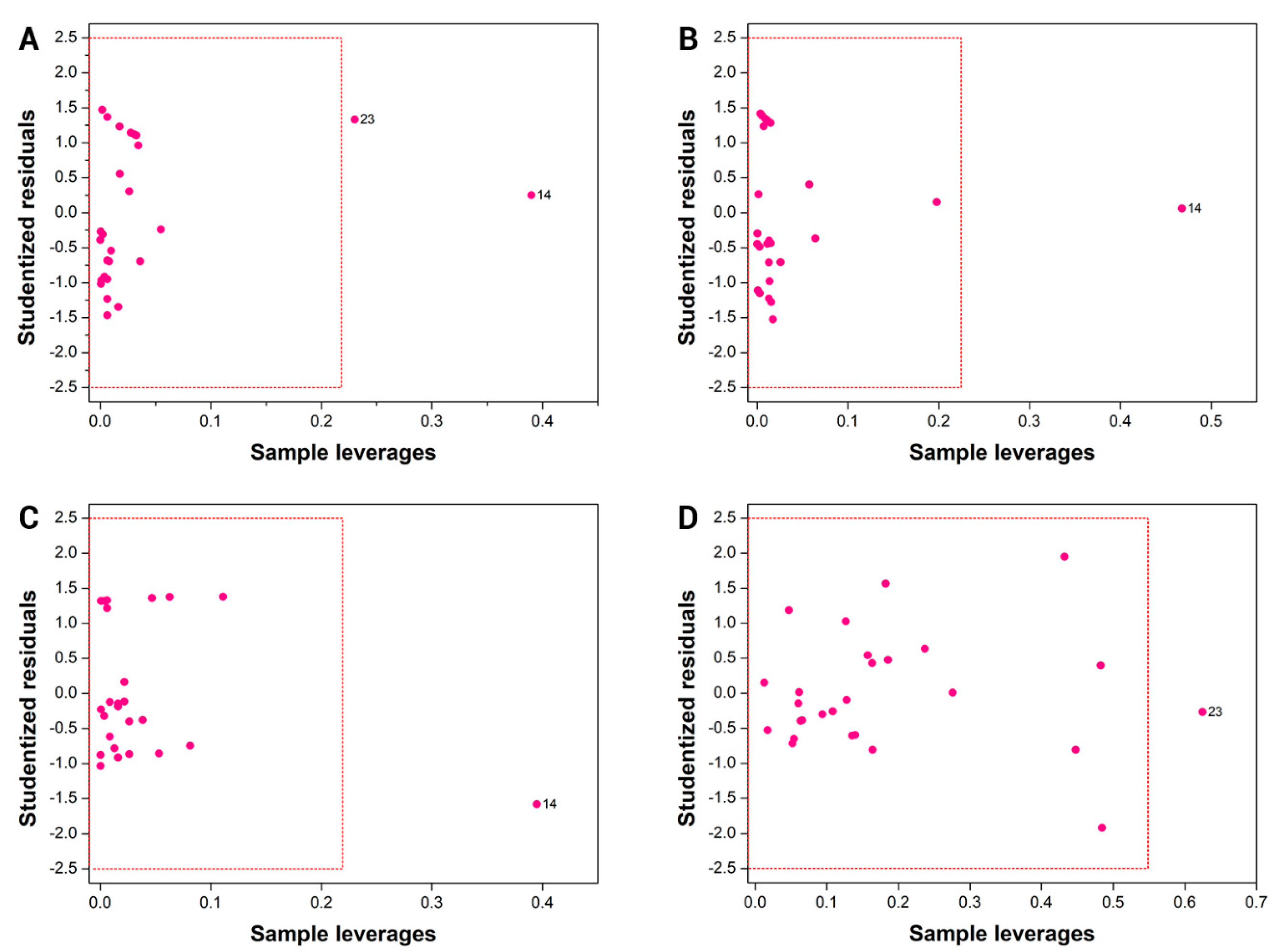
3.1. HQSAR and AutoQSAR Models
The initial HQSAR models were developed using all available hologram lengths (53 to 401 bins) and fragment sizes varying from 4 to 7 atoms. The following combinations of fragment distinction parameters were used to generate the molecular holograms: A/B/C, A/B/C/Ch, A/B/C/H, and A/B/C/H/Ch. The results for cross-validated LOO and full models are shown in
Table 2. Since significant results were obtained for all models, we evaluated the leverage of fragment size on the statistical consistency while keeping the same fragment distinction criteria. The best models obtained with this procedure are listed in
Table 3. Additionally,
Table 3 shows the
r2pred values, which confirm the predictive ability of these models for the test set.
Table 3 shows that variation in fragment size slightly improved the key statistical indicator
q2. Among the five HQSAR analyses in
Table 3, model 6 exhibited the most significant results (
q2 = 0.71 and
r2pred = 0.80). To assess the susceptibility of model 6 to chance correlations, a Y-randomization test was carried out. The noise introduced in the model by systematic perturbations in the dependent variable caused an expected drop in the
q2 value. The randomized model had a
Q2 = 0.42, which attests to the stability of the original QSAR analysis against chance correlations. Stable models should, additionally, yield progressive scrambling effective slopes close to unity, which is the case for model 6 (
dq2/dr2yy = 1.01). In addition to the procedures for internal validation, model 6 was assessed for its external predictive ability for the test set compounds. The good agreement between the experimental and predicted pIC
50 values along with an
r2pred of 0.80 demonstrate the high predictive power of the best HQSAR model for novel, structurally related compounds. Experimental and predicted pIC
50 values for the complete dataset are listed in
Table 4 and depicted in
Figure 3.
The AutoQSAR models were built using all available molecular fingerprints—linear, radial, molprint2D and dendritic—and 497 physicochemical and topological descriptors. Optimized combinations of a nonredundant descriptor subset and fingerprinting methods were correlated with the biological activity using the default machine learning algorithms of AutoQSAR. The statistical indicators for the best models are presented in
Table 5.
All models in
Table 5 performed well regarding both the training and test sets. The model with 72% of the compounds in the training set and dendritic fingerprint exhibited the best performance, as demonstrated by the highest score value of 0.90. Similarly, considering only the training set, this model yielded the best coefficient of determination (
r2 = 0.89). Considering only the test set, the model with a training set of 80% performed slightly better than the other models (
q2 = 0.91).
Table 4 and
Figure 3B demonstrate the suitable agreement between the experimental and predicted pIC
50 values and the high predictive power of the dendritic AutoQSAR model.
3.2. CoMFA and CoMSIA Models
CoMFA and CoMSIA models were built using aligned cruzain inhibitors, as illustrated in
Figure 2. Region focusing weighted by StDev*Coeff values from 0.3 to 0.9 and grid spacings from 0.5 to 2.0 Å were applied to derive the PLS regressions.
Table 6 shows the statistical indicators for the best 3D QSAR models. Grid spacings of 1.1 and 1.3 Å for CoMFA and CoMSIA, respectively, and a StDev*Coeff value of 0.3 produced the most statistically sound models. The PLS-LOO regression models produced
q2 values of 0.72 and 0.63 for the best CoMFA and CoMSIA analyses, respectively. Additionally, the best CoMFA and CoMSIA models exhibited
r2pred values of 0.81 and 0.73, respectively, demonstrating the predictive ability of these models for the test set. Progressive scrambling generated critical slopes (
dq2/
dr2yy) of 1.09 for CoMFA and 1.10 for CoMSIA.
Q2 values of 0.52 and 0.48 were obtained for the CoMFA and CoMSIA scrambling analyses, respectively.
Table 4 and
Figure 3C,D show the alignment between the experimental and predicted pIC
50 values for the CoMFA and CoMSIA models. Considering the agreement between the experimental and predicted biological activity for the test set, the 3D QSAR models have a suitable ability to predict the activity of novel compounds belonging to the structural class studied.
3.4. 2D Contribution Maps
The best HQSAR model was used to create color-coded 2D maps that assigned negative and positive contributions to activity for each molecular fragment.
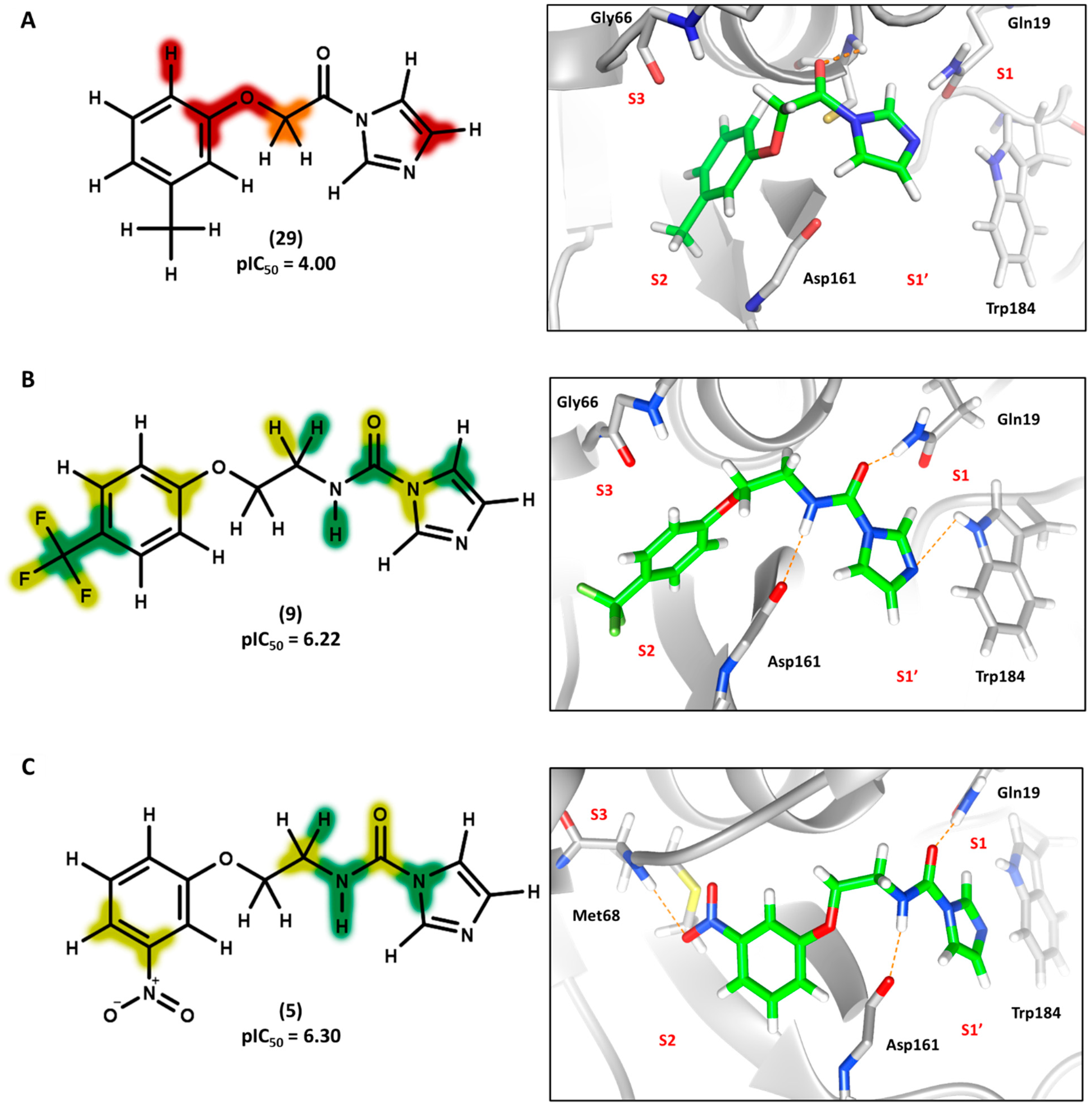
Figure 5 shows the contribution maps for the highly potent compounds 2, 3, and 6 along with their predicted binding conformations. The contribution maps indicate intermediate and positive contributions of aryl halide groups to the biological activity, corroborated by the binding mode of inhibitors 2, 3, and 6 with cruzain. The aryl halide rings of these compounds interact with the S2 subsite, which consists predominantly of hydrophobic residues. In the linker that connects the two ring systems, the amide and ether groups were flagged, playing an important role in enzyme inhibition. Indeed, the predicted binding conformations for compounds 2, 3, and 6 show the ether oxygen interacting with Gly66 and the amide hydrogen and oxygen forming hydrogen bonds with Asp161 and Gln19, respectively. The importance of the linker was further demonstrated by the negative contributions attributed to compound 29 (
Figure 6A). This compound had the linker shortened by the removal of the amide nitrogen and one of the methylene groups, which radically decreased its potency. This reduction in activity probably occurred because of the loss of key hydrogen bonds with Gln19 and Asp161. Additionally, the shortening of the linker hindered the proper positioning of the two rings in the S1′ and S2 subsites, which hampered the key elements that support cruzain-inhibitor binding.
Keeping the optimum linker configuration and varying the substitution pattern at the phenyl ring revealed further intermediate and positive contributions to activity (
Figure 6B,C). The molecular docking results for compound 9 (
Figure 6B) confirmed that hydrophobic moieties enable full interaction with the S2 subsite and represent an important driver of enzyme-inhibitor interactions. Otherwise, the polar nitro group of inhibitor 5 (
Figure 6C) allows the phenyl ring to access the S2 subsite by establishing a hydrogen bond with Met68, demonstrating that specific polar interactions can also mediate a proper interaction with the S2 subsite.
For the imidazole ring, most compounds in the dataset exhibited positive contributions for this group (
Figure 5 and
Figure 6). Imidazole was found to interact with Trp184, which plays an important role in anchoring the inhibitors at the solvent-exposed S1/S1′ interface. Replacing the imidazole with rings such as pyrimidine, pyridine, and piperidine severely decreased activity, as shown by the generalized negative contributions shown in
Figure 7.
3.5. 3D Contour Maps
3D QSAR contour maps were used to assign the leverage of steric, electrostatic, and hydrogen-bond donor/acceptor features on the activity against cruzain. CoMFA and CoMSIA green steric maps designate areas where bulky groups are associated with increased biological activity. Yellow contours, otherwise, indicate regions where the introduction of bulky moieties correlates with reduced activity. Regarding the electrostatic maps, red and blue contours designate areas where the increment in negative and positive charge, respectively, correlates with increased biological activity.
Figure 8 depicts CoMFA and CoMSIA contour maps for the lead compound (1, pIC
50 = 6.00).
Figure 8A shows the phenyl ring surrounded by green contours, which is aligned with the role played by bulky groups in the interaction with the S2 subsite, a feature that was also highlighted by the 2D contribution maps (
Figure 5 and
Figure 6). Yellow plots around the linker support the concept that the introduction of bulky substituents in this region is detrimental to activity. Bulky groups could disrupt the hydrogen bonds between the linker and Gln19 and Asp161. These residues border a relatively narrow channel that connects the two largest cavities formed by the S1′ and S2 subsites (
Figure 8E). Additionally, the electrostatic maps show red areas surrounding the amide oxygen and the imidazole nitrogen, highlighting the importance of negative dipoles in these regions. These findings are supported by the interaction of the amide oxygen and imidazole nitrogen with Gln19 and Trp184, respectively. Moreover, the blue maps next to the amide and imidazole hydrogens suggest that increasing the positive charge at these areas could enhance the activity of the compounds.
The CoMSIA electrostatic maps (
Figure 8B) reinforce the favorable role played by the negative dipole at the imidazole 3-nitrogen and the positive partial charge at the amide and imidazole hydrogen atoms. Furthermore, the CoMSIA electrostatic maps stress the relevance of the negative partial charge next to the phenyl ring, as indicated by the red contour surrounding the ether oxygen. The ether oxygen of several compounds was predicted to interact with Gly66 at the S3 subsite (
Figure 5). CoMSIA hydrogen bond donor and acceptor contour plots are pictured in
Figure 8C,D, respectively. Cyan and purple spots near the amide hydrogen and imidazole 3-nitrogen show that hydrogen bond donors are favorable and detrimental to biological activity, respectively. This is consistent with the binding mode of compound 1 (
Figure 8E), which shows a hydrogen bond between the amide hydrogen and Asp161 and the imidazole 3-nitrogen acting as an acceptor. The importance of the hydrogen-bond acceptor characteristic of imidazole is additionally stressed by the magenta contour shown in
Figure 8D. The interaction of the imidazole 3-nitrogen with Trp184 was revealed to be highly relevant to cruzain inhibition. This interaction can be responsible for stabilizing the imidazole group at the large and solvent-exposed S1/S1′ interface, providing a key enthalpic drive for cruzain-inhibitor binding. Overall, the 3D QSAR contour maps highlight molecular attributes that closely correlate with the 2D QSAR contribution maps. Altogether, both the 2D and 3D QSAR models and the molecular docking results were able to disclose relevant aspects underlying cruzain inhibition and the enzyme–inhibitor interaction.
The development of QSAR models has been a valuable tool to study cruzain inhibitors, and broadly diverse techniques have been reported recently. In a 3D QSAR study, Saraiva and co-workers developed CoMFA and CoMSIA models for a series of α–keto-based cruzain inhibitors. Solid models were built with
r2pred = 0.72 for CoMFA and 0.97 for CoMSIA. Additionally, molecular dynamics simulations were carried to examine the binding mode of the inhibitors [
39]. Pauli et al. developed HQSAR, CoMFA, and CoMSIA models for a series of benzimidazole derivatives. The final models showed sound statistical parameters and proved to be predictive for the test set compounds with
r2pred of 0.65, 0.94, and 0.82 for HQSAR, CoMFA, and CoMSIA, respectively [
21]. In another study, Scotti and colleagues used multiple linear regression (MLR), the best-first algorithm, and PLS to generate QSAR models (
r2ext = 0.79) for 61 semicarbazones. The variable selection approach used in the study resulted in the extraction of a few 1D, 2D, and 3D descriptors from more than 4800 attributes [
40]. Structurally diverse cruzain inhibitors were investigated by Rosas–Jimenez and co-workers. More than 800 cruzain inhibitors were collected from the literature and QSAR models were built using the k-nearest neighbors and random forest techniques. 1D and 2D descriptors were used and the external predictive ability of the models was demonstrated by
r2ext values of 0.72 and 0.76 [
41].
As one can see from studies that have been conducted recently, different QSAR approaches can be used to produce statistically sound models with good predictive ability. Our models are comparable with these studies in terms of statistical robustness and predictive power. Furthermore, the QSAR effort described here can contribute to the advance of the field regarding the following aspects. In this work, we report QSAR models for reversible, competitive, and non-peptidic cruzain inhibitors belonging to the imidazole class. These compounds showed promising properties including in vivo efficacy against an animal model of Chagas disease and no acute toxicity. In addition, the initial hit was discovered in a virtual screening and the analogues were optimized by extensive computational, synthetic, and biological studies entirely conducted in our laboratory.
Although the QSAR models described here were able to predict accurately the activity of the test set compounds and identify features that are determinant for cruzain inhibition, every QSAR method has advantages and limitations. 2D techniques such as AutoQSAR and HQSAR have the important advantage of being independent of molecular alignment, which makes these methods very straightforward. This is an important feature as these techniques require significantly less time for model development. Both 2D methods are based on molecular fragments as descriptors, and this type of independent variable has been widely used [
42]. Besides molecular fragmentation, AutoQSAR computes physicochemical properties, which significantly expands the scope of molecular descriptors considered in the construction of the models in comparison with HQSAR. However, 2D methods have important limitations. The main disadvantage is that three-dimensional properties that determine the interaction with molecular targets are not considered. No insight into ligand–receptor interaction has been provided by taking the output of 2D methods alone.
Otherwise, 3D methods such as CoMFA and CoMSIA allow the interpretation of the results in terms of molecular features that are closely related to ligand–receptor interactions. Features such as electrostatic and steric potentials and hydrogen-bonding ability are especially useful to presume which type of intermolecular interactions can take place during the formation of the ligand–receptor complex. When the structure of the molecular target is known, it can be integrated to the output of 3D QSAR models to give a much more detailed picture of ligand–receptor molecular recognition. An important disadvantage of 3D methods is the dependence on structural alignment, which can significantly slow the process of model development. Additionally, defining a method for structural alignment is not straightforward, mainly when the molecular target is unknown. One key difference in the output of the 3D methods used here is that CoMFA maps are more fragmented and apart from the ligand surfaces than CoMSIA maps [
20]. This occurs because of the cutoff parameterization of the CoMFA fields which results in a high steepness of the electrostatic and steric potentials when the probe comes too close to the molecular surfaces. Otherwise, the Gaussian function implemented in CoMSIA is significantly softer than the Lennard–Jones and Coulomb functions used in CoMFA. The use of the CoMSIA Gaussian function facilitates the interpretation of the contour maps as they become more continuous and able to fill in the region occupied by the compounds.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




































