
The Impact of the Ca2+-Independent Phospholipase A2β (iPLA2β) on Immune Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Macrophages
3. T-Cells and B-Cells
4. Immune Cells and Lipids
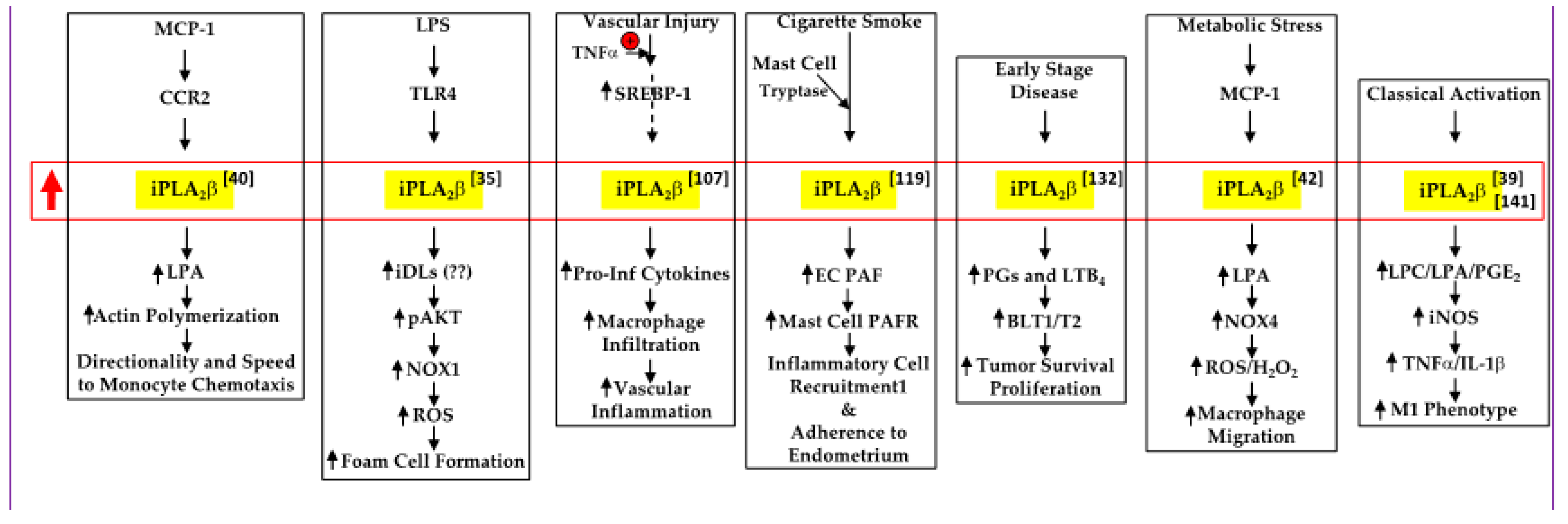
5. Deleterious Impact of iPLA2β Activation
5.1. Cellular Compass
5.2. Foam Cell Formation
5.3. Vascular Injury
5.4. Cigarette Smoke
5.5. Early-Stage Disease
5.6. Metabolic Stress
5.7. Macrophage Polarization
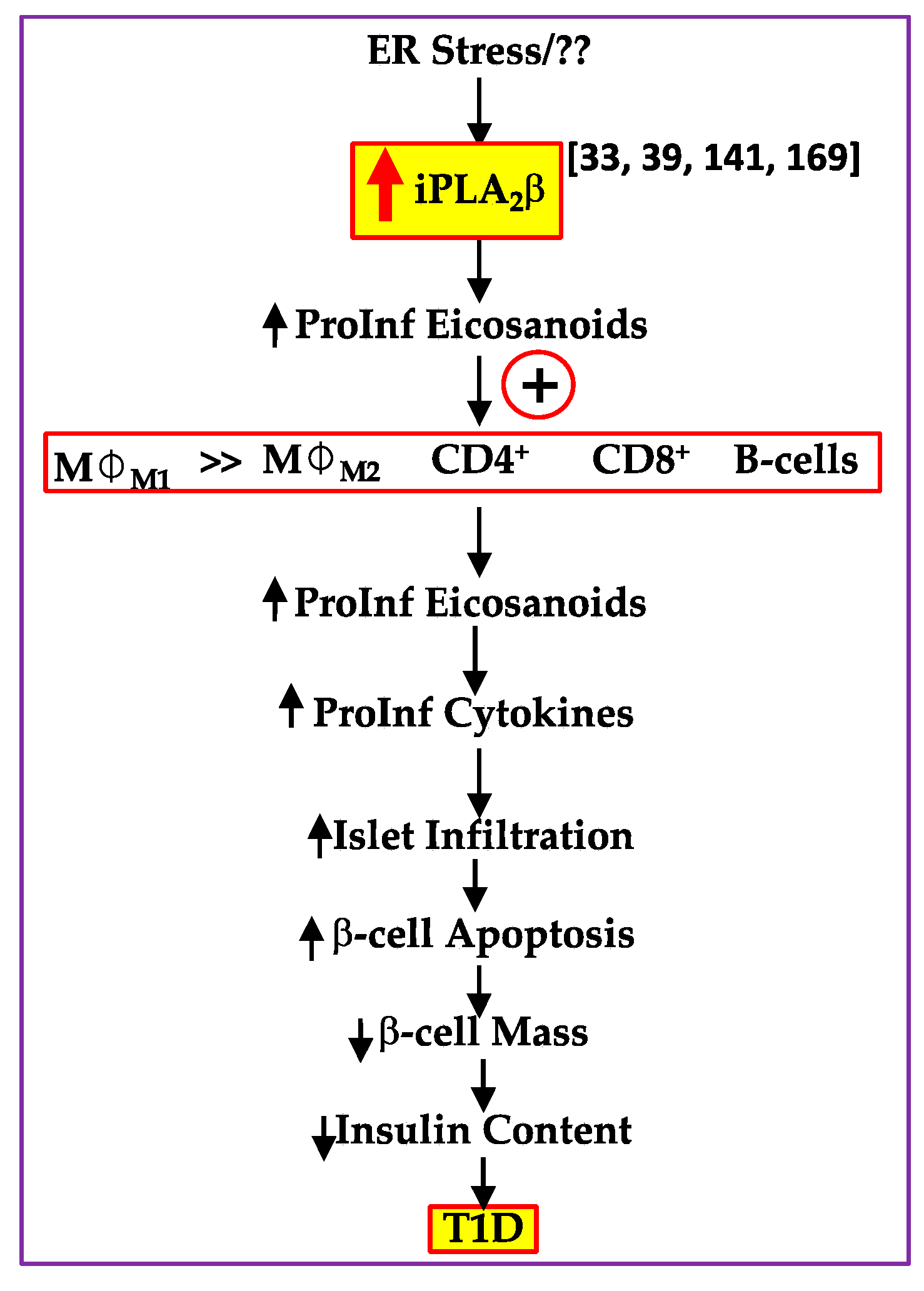
5.8. T1D Development
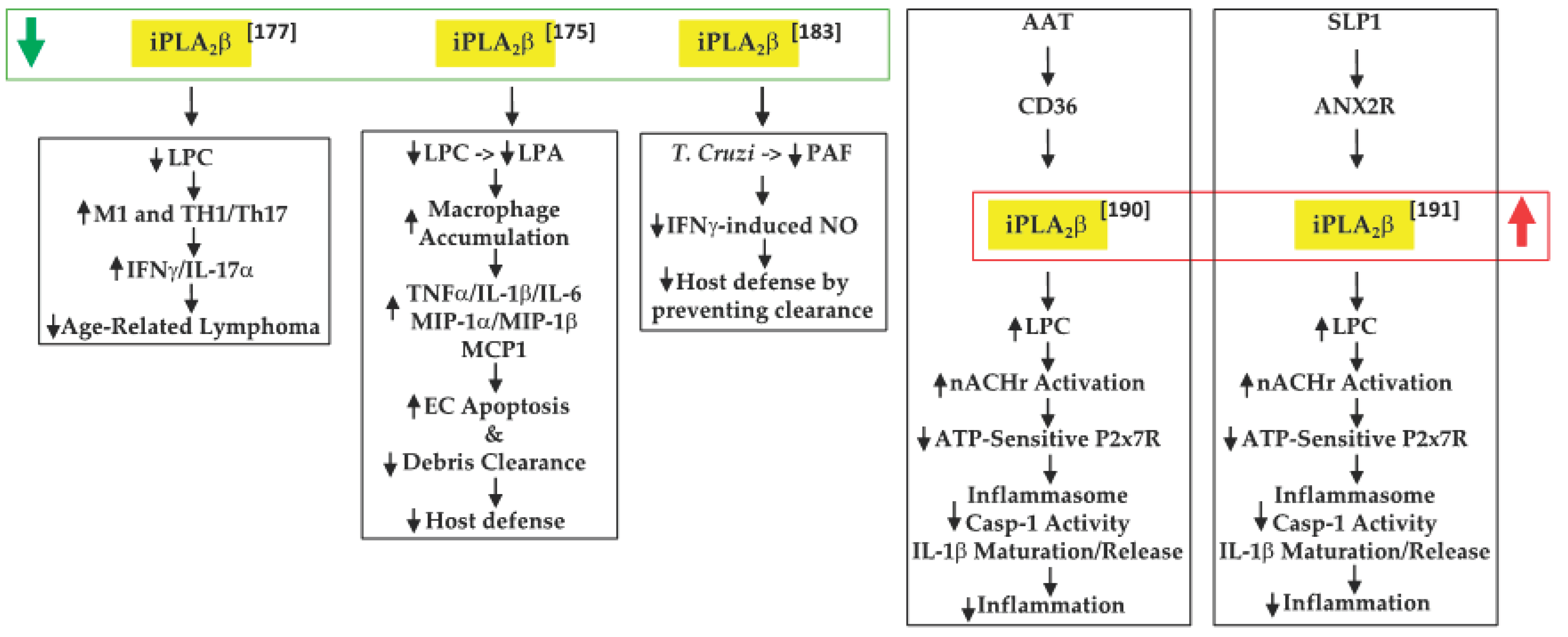
6. Protective Consequences of iPLA2β Activation
6.1. Cancer Development
6.2. Inflammatory Bowel Disease (IBD)
6.3. Chagas Disease
6.4. Negative Modulation of Inflammation by AAT and SLP1
7. Summary
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gijon, M.A.; Leslie, C.C. Phospholipases A2. Semin. Cell Dev. Biol. 1997, 8, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2b and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2beta reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ramanadham, S.; Ma, Z.A.; Bao, S.; Mancuso, D.J.; Gross, R.W.; Turk, J. Group VIA phospholipase A2 forms a signaling complex with the calcium/calmodulin-dependent protein kinase IIbeta expressed in pancreatic islet beta-cells. J. Biol. Chem. 2005, 280, 6840–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchlis, V.D.; Armando, A.; Dennis, E.A. Substrate-specific inhibition constants for phospholipase A2 acting on unique phospholipid substrates in mixed micelles and membranes using lipidomics. J. Med. Chem. 2019, 62, 1999–2007. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Chen, Y.; Mccammon, J.A.; Dennis, E.A. Membrane allostery and unique hydrophobic sites promote enzyme substrate specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, T.; Kokotos, G.; Magrioti, V.; Bone, R.N.; Mobley, J.A.; Hancock, W.; Ramanadham, S. Characterization of FKGK18 as inhibitor of group VIA Ca2+-independent phospholipase A2 (iPLA2b): Candidate drug for preventing beta-cell apoptosis and diabetes. PLoS ONE 2013, 8, e71748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokotos, G.; Hsu, Y.H.; Burke, J.E.; Baskakis, C.; Kokotos, C.G.; Magrioti, V.; Dennis, E.A. Potent and selective fluoroketone inhibitors of group VIA calcium-independent phospholipase A2. J. Med. Chem. 2010, 53, 3602–3610. [Google Scholar] [CrossRef] [Green Version]
- Gross, R.W.; Ramanadham, S.; Kruszka, K.K.; Han, X.; Turk, J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 1993, 32, 327–336. [Google Scholar] [CrossRef]
- Bao, S.; Jin, C.; Zhang, S.; Turk, J.; Ma, Z.; Ramanadham, S. Beta-cell calcium-independent group VIA phospholipase A2 (iPLA2b): Tracking iPLA2b movements in response to stimulation with insulin secretagogues in INS-1 cells. Diabetes 2004, 53, S186–S189. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Zhang, S.; Bohrer, A.; Ramanadham, S. Calcium-independent phospholipase A2 (iPLA2b)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J. Biol. Chem. 2008, 283, 34819–34832. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Ramanadham, S.; Wohltmann, M.; Bohrer, A.; Hsu, F.F.; Turk, J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2b) indicate a signaling rather than a housekeeping role for iPLA2b. J. Biol. Chem. 2001, 276, 13198–13208. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhang, S.; Turk, J.; Ramanadham, S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2b in INS-1 insulinoma cells. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E820–E833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanadham, S.; Hsu, F.F.; Zhang, S.; Jin, C.; Bohrer, A.; Song, H.; Bao, S.; Ma, Z.; Turk, J. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2b) and suppressed by inhibition of iPLA2b. Biochemistry 2004, 43, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2b. Biochim. Biophys. Acta 2010, 1801, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanadham, S.; Hsu, F.F.; Bohrer, A.; Zhongmin, M.; Turk, J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J. Biol. Chem. 1999, 275, 13915–13927. [Google Scholar] [CrossRef] [Green Version]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The crucial roles of inflammatory mediators in inflammation: A review. Vet. World 2018, 11, 627–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanapure, S.P.; Garvey, D.S.; Janero, D.R.; Letts, L.G. Eicosanoids in inflammation: Biosynthesis, pharmacology, and therapeutic frontiers. Curr. Top. Med. Chem. 2007, 7, 311–340. [Google Scholar] [CrossRef]
- Luo, P.; Wang, M.H. Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat. 2011, 95, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tessaro, F.H.; Ayala, T.S.; Martins, J.O. Lipid mediators are critical in resolving inflammation: A review of the emerging roles of eicosanoids in diabetes mellitus. Biomed. Res. Int. 2015, 2015, 568408. [Google Scholar] [CrossRef] [Green Version]
- Umamaheswaran, S.; Dasari, S.K.; Yang, P.; Lutgendorf, S.K.; Sood, A.K. Stress, inflammation, and eicosanoids: An emerging perspective. Cancer Metastasis Rev. 2018, 37, 203–211. [Google Scholar] [CrossRef]
- Zaitseva, L.; Vaisburd, M.; Shaposhnikova, G.; Mysyakin, E. Role of eicosanoids in regulation of macrophage phagocytic functions by platelet-activating factor during endotoxic shock. Bull. Exp. Biol. Med. 2000, 130, 879–881. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; O’donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356. [Google Scholar] [CrossRef] [PubMed]
- Issan, Y.; Hochhauser, E.; Guo, A.; Gotlinger, K.H.; Kornowski, R.; Leshem-Lev, D.; Lev, E.; Porat, E.; Snir, E.; Thompson, C.I.; et al. Elevated level of pro-inflammatory eicosanoids and EPC dysfunction in diabetic patients with cardiac ischemia. Prostaglandins Other Lipid Mediat. 2013, 100–101, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Gilroy, D.W.; Newson, J.; Sawmynaden, P.; Willoughby, D.A.; Croxtall, J.D. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. FASEB J. 2004, 18, 489–498. [Google Scholar] [CrossRef]
- De Jong, A.J.; Kloppenburg, M.; Toes, R.E.; Ioan-Facsinay, A. Fatty acids, lipid mediators, and T-cell function. Front. Immunol. 2014, 5, 483. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bone, R.N.; Gai, Y.; Magrioti, V.; Kokotou, M.G.; Ali, T.; Lei, X.; Tse, H.M.; Kokotos, G.; Ramanadham, S. Inhibition of Ca2+-independent phospholipase A2beta (iPLA2b) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 2015, 64, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-De-Gomez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J. Immunol. 2014, 192, 752–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Park, D.W.; Park, S.C.; Park, Y.K.; Hong, S.Y.; Kim, J.R.; Lee, C.H.; Baek, S.H. Calcium-independent phospholipase A2beta-Akt signaling is involved in lipopolysaccharide-induced NADPH oxidase 1 expression and foam cell formation. J. Immunol. 2009, 183, 7497–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Li, Y.; Lei, X.; Wohltmann, M.; Jin, W.; Bohrer, A.; Semenkovich, C.F.; Ramanadham, S.; Tabas, I.; Turk, J. Attenuated free cholesterol loading-induced apoptosis but preserved phospholipid composition of peritoneal macrophages from mice that do not express group VIA phospholipase A2. J. Biol. Chem. 2007, 282, 27100–27114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Bohrer, A.; Wohltmann, M.; Ramanadham, S.; Hsu, F.F.; Turk, J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids 2001, 36, 689–700. [Google Scholar] [CrossRef]
- Song, Y.; Wilkins, P.; Hu, W.; Murthy, K.S.; Chen, J.; Lee, Z.; Oyesanya, R.; Wu, J.; Barbour, S.E.; Fang, X. Inhibition of calcium-independent phospholipase A2 suppresses proliferation and tumorigenicity of ovarian carcinoma cells. Biochem. J. 2007, 406, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of macrophages toward M2 phenotype is favored by reduction in iPLA2b (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.S.; Carnevale, K.A.; Cathcart, M.K. iPLA2beta: Front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 2008, 205, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Ayilavarapu, S.; Kantarci, A.; Fredman, G.; Turkoglu, O.; Omori, K.; Liu, H.; Iwata, T.; Yagi, M.; Hasturk, H.; Van Dyke, T.E. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J. Immunol. 2010, 184, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Day, R.; Bao, S.; Turk, J.; Zhao, Q.D. Group VIA phospholipase A2 mediates enhanced macrophage migration in diabetes mellitus by increasing expression of nicotinamide adenine dinucleotide phosphate oxidase 4. Arter. Thromb. Vasc. Biol. 2014, 34, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyvas, A.; Baskakis, C.; Magrioti, V.; Constantinou-Kokotou, V.; Stephens, D.; Lopez-Vales, R.; Lu, J.Q.; Yong, V.W.; Dennis, E.A.; Kokotos, G.; et al. Differing roles for members of the phospholipase A2 superfamily in experimental autoimmune encephalomyelitis. Brain J. Neurol. 2009, 132, 1221–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mchowat, J.; Gullickson, G.; Hoover, R.G.; Sharma, J.; Turk, J.; Kornbluth, J. Platelet-activating factor and metastasis: Calcium-independent phospholipase A2beta deficiency protects against breast cancer metastasis to the lung. Am. J. Physiol. Cell Physiol. 2011, 300, C825–C832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotera, T.M.; Schuster, D.P.; Bourhim, M.; Chadha, K.; Klaich, G.; Corral, D.A. Regulation of PSA secretion and survival signaling by calcium-independent phopholipase A2beta in prostate cancer cells. Prostate 2009, 69, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, M.R.; Anfuso, C.D.; Lupo, G.; Motta, C.; Romeo, L.; Guerra, L.; Cappellani, A.; Ragusa, N.; Cantarella, G.; Alberghina, M. Expression of Ca2+-independent and Ca2+-dependent phospholipases A2 and cyclooxygenases in human melanocytes and malignant melanoma cell lines. Biochim. Biophys. Acta 2008, 1781, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Thayer, T.C.; Delano, M.; Liu, C.; Chen, J.; Padgett, L.E.; Tse, H.M.; Annamali, M.; Piganelli, J.D.; Moldawer, L.L.; Mathews, C.E. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes 2011, 60, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Jiang, J.X.; Zhang, G.X. Macrophages: A double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 2014, 160, 17–22. [Google Scholar] [CrossRef]
- Burg, A.R.; Tse, H.M. Redox-sensitive innate immune pathways during macrophage activation in type 1 diabetes. Antioxid. Redox Signal. 2018, 29, 1373–1398. [Google Scholar] [CrossRef]
- Hirayama, D.; Iida, T.; Nakase, H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [Green Version]
- Schenten, D.; Medzhitov, R. The control of adaptive immune responses by the innate immune system. Adv. Immunol. 2011, 109, 87–124. [Google Scholar] [PubMed]
- Colbert, J.D.; Cruz, F.M.; Rock, K.L. Cross-presentation of exogenous antigens on MHC I molecules. Curr. Opin. Immunol. 2020, 64, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Theret, M.; Mounier, R.; Rossi, F. The origins and non-canonical functions of macrophages in development and regeneration. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; Van Rooijen, N.; et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, A.; Strack, E.; Snodgrass, R.G.; Brüne, B. mPGES-1 and ALOX5/-15 in tumor-associated macrophages. Cancer Metastasis Rev. 2018, 37, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Pro-resolving mediators in regulating and conferring macrophage function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef] [Green Version]
- Norris, P.C.; Dennis, E.A. A lipidomic perspective on inflammatory macrophage eicosanoid signaling. Adv. Biol. Regul. 2014, 54, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Bi, X.; Wang, S.; Zhang, Z.; Li, F.; Zhao, A.Z. Therapeutic Potential of ω-3 polyunsaturated fatty acids in human autoimmune diseases. Front. Immunol. 2019, 10, 2241. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Gronert, K. Eicosanoid and specialized proresolving mediator regulation of lymphoid Cells. Trends Biochem. Sci. 2019, 44, 214–225. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M. Generation of Lymphocytes in Bone Marrow and Thymus; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 5, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. B cell development, activation and effector functions. Primer Immune Response 2014, 2, 111–142. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef]
- Hooper, K.M.; Kong, W.; Ganea, D. Prostaglandin E2 inhibits Tr1 cell differentiation through suppression of c-Maf. PLoS ONE 2017, 12, e0179184. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Frosen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E2-EP2-NF-kappaB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, V.; Gurlo, T.; Jarstadmarken, H.O.; Von Grafenstein, H. Regulation of TCR-induced IFN-gamma release from islet-reactive non-obese diabetic CD8+ T cells by prostaglandin E2 receptor signaling. Int. Immunol. 2000, 12, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.J.; Sun, Y.J.; Zhu, D.Y.; Chen, Q.; Han, X. Potential role of NO in modulation of COX-2 expression and PGE2 production in pancreatic beta-cells. Acta Biochim. Biophys. Sin. 2005, 37, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boizel, R.; Bruttmann, G.; Benhamou, P.Y.; Halimi, S.; Stanke-Labesque, F. Regulation of oxidative stress and inflammation by glycaemic control: Evidence for reversible activation of the 5-lipoxygenase pathway in type 1, but not in type 2 diabetes. Diabetologia 2010, 53, 2068–2070. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.K.; Cole, B.K.; Wen, Y.; Keller, S.R.; Nadler, J.L. 12/15-lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity 2009, 17, 1657–1663. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Taylor-Fishwick, D.A.; Nadler, J.L. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia 2016, 59, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, J.A.; Li, S.D.; Yang, A.; Huang, L.; Kole, R. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res. 2010, 38, 8348–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inceoglu, B.; Bettaieb, A.; Haj, F.G.; Gomes, A.V.; Hammock, B.D. Modulation of mitochondrial dysfunction and endoplasmic reticulum stress are key mechanisms for the wide-ranging actions of epoxy fatty acids and soluble epoxide hydrolase inhibitors. Prostaglandins Other Lipid Mediat. 2017, 133, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Hwang, H.J.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Baik, S.H.; Choi, K.M. Resolvin D1 reduces ER stress-induced apoptosis and triglyceride accumulation through JNK pathway in HepG2 cells. Mol. Cell. Endocrinol. 2014, 391, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, J.; Wang, M.H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2016, 125, 80–89. [Google Scholar] [CrossRef]
- Jouihan, S.A.; Zuloaga, K.L.; Zhang, W.; Shangraw, R.E.; Krasnow, S.M.; Marks, D.L.; Alkayed, N.J. Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J. Cereb. Blood Flow Metab. 2013, 33, 1650–1656. [Google Scholar] [CrossRef]
- Luther, J.M.; Brown, N.J. Epoxyeicosatrienoic acids and glucose homeostasis in mice and men. Prostaglandins Other Lipid Mediat. 2016, 125, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Clare-Salzler, M. Eicosanoid imbalance in the NOD mouse is related to a dysregulation in soluble epoxide hydrolase and 15-PGDH expression. Ann. N. Y. Acad. Sci. 2006, 1079, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol. Ther. 2005, 4, 924–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, M.A. Mechanisms underlying the loss of self tolerance in NOD mice. Res. Immunol. 1997, 148, 301–306. [Google Scholar] [CrossRef]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Ming, W.J.; Bersani, L.; Mantovani, A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J. Immunol. 1987, 138, 1469–1474. [Google Scholar]
- Ma, K.; Nunemaker, C.S.; Wu, R.; Chakrabarti, S.K.; Taylor-Fishwick, D.A.; Nadler, J.L. 12-Lipoxygenase products reduce insulin secretion and {beta}-cell viability in human islets. J. Clin. Endocrinol. Metab. 2010, 95, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Mcduffie, M.; Maybee, N.A.; Keller, S.R.; Stevens, B.K.; Garmey, J.C.; Morris, M.A.; Kropf, E.; Rival, C.; Ma, K.; Carter, J.D.; et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 2008, 57, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.M.; Thimmalapura, P.R.; Woode, E.A.; Nadler, J.L. Evidence that increased 12-lipoxygenase expression impairs pancreatic beta cell function and viability. Biochem. Biophys. Res. Commun. 2003, 308, 427–432. [Google Scholar] [CrossRef]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Gu, J.; Vandenhoff, G.E.; Liu, X.; Nadler, J.L. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1933–H1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.H.; Pesant, S.; Cohn, H.I.; Eckhart, A.D. Enhanced sterol response element-binding protein in postintervention restenotic blood vessels plays an important role in vascular smooth muscle proliferation. Life Sci. 2008, 82, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Bleich, D.; Chen, S.; Zipser, B.; Sun, D.; Funk, C.D.; Nadler, J.L. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J. Clin. Investig. 1999, 103, 1431–1436. [Google Scholar] [CrossRef] [Green Version]
- Green-Mitchell, S.M.; Tersey, S.A.; Cole, B.K.; Ma, K.; Kuhn, N.S.; Cunningham, T.D.; Maybee, N.A.; Chakrabarti, S.K.; Mcduffie, M.; Taylor-Fishwick, D.A.; et al. Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS ONE 2013, 8, e56763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzesik, W.J.; Nadler, J.L.; Machida, Y.; Nadler, J.L.; Imai, Y.; Morris, M.A. Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E387–E395. [Google Scholar] [CrossRef]
- Bennett, M.; Gilroy, D.W. Lipid mediators in inflammation. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Tuosto, L.; Xu, C. Editorial: Membrane lipids in T cell functions. Front. Immunol. 2018, 9, 1608. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Tseng, S.C.; Rollins, B.J. Monocyte chemoattractant protein-1. Chem. Immunol. 1999, 72, 7–29. [Google Scholar] [PubMed]
- Cathcart, M.K. Signal-activated phospholipase regulation of leukocyte chemotaxis. J. Lipid Res. 2009, 50, S231–S236. [Google Scholar] [CrossRef] [Green Version]
- Kam, Y.; Exton, J.H. Phospholipase D activity is required for actin stress fiber formation in fibroblasts. Mol. Cell. Biol. 2001, 21, 4055–4066. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kriz, R.W.; Wolfman, N.; Shaffer, M.; Seehra, J.; Jones, S.S. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 1997, 272, 8567–8575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, C.M.; Han, X.; Mancuso, D.J.; Gross, R.W. Identification of calcium-independent phospholipase A2 (iPLA2) beta, and not iPLA2gamma, as the mediator of arginine vasopressin-induced arachidonic acid release in A-10 smooth muscle cells. Enantioselective mechanism-based discrimination of mammalian iPLA2s. J. Biol. Chem. 2002, 277, 32807–32814. [Google Scholar] [PubMed] [Green Version]
- Liu, S.; Xie, Z.; Zhao, Q.; Pang, H.; Turk, J.; Calderon, L.; Su, W.; Zhao, G.; Xu, H.; Gong, M.C.; et al. Smooth muscle-specific expression of calcium-independent phospholipase A2beta (iPLA2b) participates in the initiation and early progression of vascular inflammation and neointima formation. J. Biol. Chem. 2012, 287, 24739–24753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wang, D.; Zhao, Z.; Xiao, Y.; Sengupta, S.; Xiao, Y.; Zhang, R.; Lauber, K.; Wesselborg, S.; Feng, L.; et al. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J. Biol. Chem. 2006, 281, 29357–29368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.G.; Lim, E.J.; Park, D.W.; Lee, S.H.; Kim, J.R.; Baek, S.H. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal. 2008, 20, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Weyand, C.M. Macrophages in vascular inflammation. From atherosclerosis to vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Zhang, S.; Barbour, S.E.; Bohrer, A.; Ford, E.L.; Koizumi, A.; Papa, F.R.; Ramanadham, S. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: A role for regulation by SREBP-1. J. Biol. Chem. 2010, 285, 6693–6705. [Google Scholar] [CrossRef] [Green Version]
- Seashols, S.J.; Del Castillo Olivares, A.; Gil, G.; Barbour, S.E. Regulation of group VIA phospholipase A2 expression by sterol availability. Biochim. Biophys. Acta 2004, 1684, 29–37. [Google Scholar] [CrossRef]
- Smani, T.; Zakharov, S.I.; Csutora, P.; Leno, E.; Trepakova, E.S.; Bolotina, V.M. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell. Biol. 2004, 6, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Wang, J.; Turk, J.; Gross, R.W. Depletion of intracellular calcium stores activates smooth muscle cell calcium-independent phospholipase A2. A novel mechanism underlying arachidonic acid mobilization. J. Biol. Chem. 1997, 272, 1522–1526. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Bone, R.N.; Ali, T.; Wohltmann, M.; Gai, Y.; Goodwin, K.J.; Bohrer, A.E.; Turk, J.; Ramanadham, S. Genetic modulation of islet beta-cell iPLA2b expression provides evidence for its impact on beta-cell apoptosis and autophagy. Islets 2013, 5, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Bone, R.N.; Ali, T.; Zhang, S.; Bohrer, A.; Tse, H.M.; Bidasee, K.R.; Ramanadham, S. Evidence of contribution of iPLA2b-mediated events during islet beta-cell apoptosis due to proinflammatory cytokines suggests a role for iPLA2b in T1D development. Endocrinology 2014, 155, 3352–3364. [Google Scholar] [CrossRef] [PubMed]
- Marentette, J.; Kolar, G.; Mchowat, J. Increased susceptibility to bladder inflammation in smokers: Targeting the PAF-PAF receptor interaction to manage inflammatory cell recruitment. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Filippou, A.; Sant, G.; Theoharides, T.C. Expression of intercellular adhesion molecules in the bladder of patients with interstitial cystitis. Urology 2004, 63, 688–693. [Google Scholar] [CrossRef]
- Rastogi, P.; White, M.C.; Rickard, A.; Mchowat, J. Potential mechanism for recruitment and migration of CD133 positive cells to areas of vascular inflammation. Thromb. Res. 2008, 123, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.; Portell, C.; Kell, P.J.; Vinson, S.M.; Mchowat, J. Protease-activated receptor stimulation activates a Ca2+-independent phospholipase A2 in bladder microvascular endothelial cells. Am. J. Physiol. Renal Physiol. 2005, 288, F714–F721. [Google Scholar] [CrossRef]
- Ueno, N.; Taketomi, Y.; Yamamoto, K.; Hirabayashi, T.; Kamei, D.; Kita, Y.; Shimizu, T.; Shinzawa, K.; Tsujimoto, Y.; Ikeda, K.; et al. Analysis of two major intracellular phospholipases A2 (PLA2) in mast cells reveals crucial contribution of cytosolic PLA2alpha, not Ca2+-independent PLA2beta, to lipid mobilization in proximal mast cells and distal fibroblasts. J. Biol. Chem. 2011, 286, 37249–37263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, D.A.; Fu, W.; Schonefeldt, S.; Feyerabend, T.B.; Ortiz-Lopez, A.; Lampi, Y.; Liston, A.; Mathis, D.; Rodewald, H.R. Type 1 diabetes in NOD mice unaffected by mast cell deficiency. Diabetes 2014, 63, 3827–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, L.; Masini, M.; Bugliani, M.; Marselli, L.; Suleiman, M.; Boggi, U.; Nogueira, T.C.; Filipponi, F.; Occhipinti, M.; Campani, D.; et al. Mast cells infiltrate pancreatic islets in human type 1 diabetes. Diabetologia 2015, 58, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.P.; Pollock, S.J.; Kaur, K.; Felgar, R.E.; Bernstein, S.H.; Chiorazzi, N.; Phipps, R.P. Constitutive and activation-inducible cyclooxygenase-2 expression enhances survival of chronic lymphocytic leukemia B cells. Clin. Immunol. 2006, 120, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Barbarotto, E.; Gonelli, A.; Tiribelli, M.; Zerbinati, C.; Celeghini, C.; Agostinelli, C.; Pileri, S.A.; Zauli, G. Potential pathogenetic implications of cyclooxygenase-2 overexpression in B chronic lymphoid leukemia cells. Am. J. Pathol. 2005, 167, 1599–1607. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.M.; Akbiyik, F.; Bernstein, S.H.; Phipps, R.P. CD40 engagement prevents peroxisome proliferator-activated receptor gamma agonist-induced apoptosis of B lymphocytes and B lymphoma cells by an NF-kappaB-dependent mechanism. J. Immunol. 2005, 174, 4060–4069. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Roshak, A.K.; Capper, E.A.; Stevenson, C.; Eichman, C.; Marshall, L.A. Human calcium-independent phospholipase A2 mediates lymphocyte proliferation. J. Biol. Chem. 2000, 275, 35692–35698. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.I.; Singh, J.; Niknami, M.; Kurek, C.; Yao, M.; Lu, S.; Maclean, F.; King, N.J.; Gelb, M.H.; Scott, K.F.; et al. Cytosolic phospholipase A2-alpha: A potential therapeutic target for prostate cancer. Clin. Cancer Res. 2008, 14, 8070–8079. [Google Scholar] [CrossRef] [Green Version]
- Guriec, N.; Le Jossic-Corcos, C.; Simon, B.; Ianotto, J.C.; Tempescul, A.; Dreano, Y.; Salaun, J.P.; Berthou, C.; Corcos, L. The arachidonic acid-LTB4-BLT2 pathway enhances human B-CLL aggressiveness. Biochim. Biophys. Acta 2014, 1842, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Qiao, M.; Schroder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullevig, S.; Zhao, Q.; Lee, C.F.; Seok Kim, H.; Zamora, D.; Asmis, R. NADPH oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Zhang, S.; Bohrer, A.; Bao, S.; Song, H.; Ramanadham, S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 2007, 46, 10170–10185. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.J.; Stephenson, D.J.; Cardona, C.L.; Lei, X.; Almutairi, A.; White, T.D.; Tusing, Y.G.; Park, M.A.; Barbour, S.E.; Chalfant, C.E.; et al. Macrophage polarization is linked to Ca2+-independent phospholipase A2beta-derived lipids and cross-cell signaling in mice. J. Lipid Res. 2020, 61, 143–158. [Google Scholar] [CrossRef]
- Gil-De-Gomez, L.; Monge, P.; Rodriguez, J.P.; Astudillo, A.M.; Balboa, M.A.; Balsinde, J. Phospholipid arachidonic acid remodeling during phagocytosis in mouse peritoneal macrophages. Biomedicines 2020, 8, 274. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.; Daka, A.; Lamour, N.F.; Mashiach, R.; Glucksam, Y.; Meijler, M.M.; Chalfant, C.E.; Zor, T. A ceramide analog inhibits cPLA2 activity and consequent PGE2 formation in LPS-stimulated macrophages. Immunol. Lett. 2011, 135, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Naraba, H.; Murakami, M.; Matsumoto, H.; Shimbara, S.; Ueno, A.; Kudo, I.; Oh-Ishi, S. Segregated coupling of phospholipases A2, cyclooxygenases, and terminal prostanoid synthases in different phases of prostanoid biosynthesis in rat peritoneal macrophages. J. Immunol. 1998, 160, 2974–2982. [Google Scholar]
- Qi, H.Y.; Shelhamer, J.H. Toll-like receptor 4 signaling regulates cytosolic phospholipase A2 activation and lipid generation in lipopolysaccharide-stimulated macrophages. J. Biol. Chem. 2005, 280, 38969–38975. [Google Scholar] [CrossRef] [Green Version]
- Ruiperez, V.; Casas, J.; Balboa, M.A.; Balsinde, J. Group V phospholipase A2-derived lysophosphatidylcholine mediates cyclooxygenase-2 induction in lipopolysaccharide-stimulated macrophages. J. Immunol. 2007, 1790, 631–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Zhou, J.; Li, T.; Mu, J.; Jin, L.; Li, S. Urocortin participates in LPS-induced apoptosis of THP-1 macrophages via S1P-cPLA2 signaling pathway. Eur. J. Pharmacol. 2020, 887, 173559. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Role of immune system in type 1 diabetes mellitus pathogenesis. Int. Immunopharmacol. 2014, 22, 182–191. [Google Scholar] [CrossRef]
- Mathis, D.; Vence, L.; Benoist, C. Beta-cell death during progression to diabetes. Nature 2001, 414, 792–798. [Google Scholar] [CrossRef]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Padgett, L.E.; Broniowska, K.A.; Hansen, P.A.; Corbett, J.A.; Tse, H.M. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N. Y. Acad. Sci. 2013, 1281, 16–35. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon, B.; Suri, A.; Unanue, E.R. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: Studies from an acute model. Am. J. Pathol. 2006, 169, 2137–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsa, R.; Andresen, P.; Gillett, A.; Mia, S.; Zhang, X.M.; Mayans, S.; Holmberg, D.; Harris, R.A. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 2012, 61, 2881–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharov, P.N.; Hu, H.; Wan, X.; Unanue, E.R. Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Unanue, E.R. Macrophages in endocrine glands, with emphasis on pancreatic islets. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Calderon, B.; Suri, A.; Miller, M.J.; Unanue, E.R. Dendritic cells in islets of Langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 6121–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, J.F.; Levisetti, M.G.; Calderon, B.; Herzog, J.W.; Petzold, S.J.; Unanue, E.R. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat. Immunol. 2010, 11, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Feduska, J.M.; Tse, H.M. The proinflammatory effects of macrophage-derived NADPH oxidase function in autoimmune diabetes. Free Radic. Biol. Med. 2018, 125, 81–89. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-mediated beta cell destruction: Autoimmunity and alloimmunity in the context of type 1 diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Dimeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Li, J.-H.; Zhu, S.-H.; Tang, H.-T.; Xia, Z.-F. Macrophage migration inhibitory factor counter-regulates dexamethasone-induced annexin 1 expression and influences the release of eicosanoids in murine macrophages. Immunology 2013, 140, 250–258. [Google Scholar] [CrossRef]
- Duan, L.; Gan, H.; Arm, J.; Remold, H.G. Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J. Immunol. 2001, 166, 7469–7476. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.M.; Gong, M.C.; Turk, J.; Post, S.R. Class A scavenger receptor-mediated macrophage adhesion requires coupling of calcium-independent phospholipase A2 and 12/15-lipoxygenase to Rac and Cdc42 activation. J. Biol. Chem. 2007, 282, 33405–33411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilorenzo, T.; Graser, R.; Ono, T.; Christianson, G.; Chapman, H.; Roopenian, D.; Nathenson, S.; Serreze, D. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc. Natl. Acad. Sci. USA 1998, 95, 12438–12543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahnema, P.; Shimoni, Y.; Nygren, A. Reduced conduction reserve in the diabetic rat heart: Role of iPLA2 activation in the response to ischemia. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H326–H334. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Stephenson, D.J.; Bone, R.N.; Cardona, C.L.; Park, M.A.; Tusing, Y.G.; Lei, X.; Kokotos, G.; Graves, C.L.; Mathews, C.E.; et al. Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Held, W.; Macdonald, H.R.; Weissman, I.L.; Hess, M.W.; Mueller, C. Genes encoding tumor necrosis factor alpha and granzyme A are expressed during development of autoimmune diabetes. Proc. Natl. Acad. Sci. USA 1990, 87, 2239–2243. [Google Scholar] [CrossRef] [Green Version]
- Herrera, P.L.; Harlan, D.M.; Vassalli, P. A mouse CD8 T cell-mediated acute autoimmune diabetes independent of the perforin and Fas cytotoxic pathways: Possible role of membrane TNF. Proc. Natl. Acad. Sci. USA 2000, 97, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, L.; Inhoffen, J.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Sun, Z.; Chamulitrat, W. Deficiency of group VIA phospholipase A2 (iPLA2b) renders susceptibility for chemical-induced colitis. Dig. Dis. Sci. 2015, 60, 3590–3602. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tuma, S.; Katava, N.; Pathil-Warth, A.; Stremmel, W.; Chamultrat, W. Deficiencies of calcium-independent phospholipase A2 B in vivo causes reducedsystemic lipids and lipoproteins concomitant with increased hepatic apoptosis and inflammation. In Journal of Hepathology, Proceeding of The International Liver CongressTM (EASL), Vienna, Austria, 10–14 April 2012; EASL: Barcelona, Spain, 2012. [Google Scholar]
- Inhoffen, J.; Tuma-Kellner, S.; Straub, B.; Stremmel, W.; Chamulitrat, W. Deficiency of iPLA2beta primes immune cells for proinflammation: Potential involvement in age-related mesenteric lymph node lymphoma. Cancers 2015, 7, 2427–2442. [Google Scholar] [CrossRef] [PubMed]
- Park, D.R.; Thomsen, A.R.; Frevert, C.W.; Pham, U.; Skerrett, S.J.; Kiener, P.A.; Liles, W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 2003, 170, 6209–6216. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 2004, 4, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Murase, R.; Sato, H.; Yamamoto, K.; Ushida, A.; Nishito, Y.; Ikeda, K.; Kobayashi, T.; Yamamoto, T.; Taketomi, Y.; Murakami, M. Group X secreted phospholipase A2 releases omega3 polyunsaturated fatty acids, suppresses colitis, and promotes sperm fertility. J. Biol. Chem. 2016, 291, 6895–6911. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.; Blase, J.R.; Hoft, D.F.; Marentette, J.O.; Turk, J.; Mchowat, J. Mice with genetic deletion of group via phospholipase a2beta exhibit impaired macrophage function and increased parasite load in Trypanosoma cruzi-induced myocarditis. Infect. Immun. 2016, 84, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Bern, C.; Verastegui, M.; Gilman, R.H.; Lafuente, C.; Galdos-Cardenas, G.; Calderon, M.; Pacori, J.; Del Carmen Abastoflor, M.; Aparicio, H.; Brady, M.F.; et al. Congenital Trypanosoma cruzi transmission in Santa Cruz, Bolivia. Clin. Infect. Dis. 2009, 49, 1667–1674. [Google Scholar] [CrossRef] [Green Version]
- Rassi, A., Jr.; Rassi, A.; Little, W.C. Chagas’ heart disease. Clin. Cardiol. 2000, 23, 883–889. [Google Scholar]
- Lannes-Vieira, J.; Silverio, J.C.; Pereira, I.R.; Vinagre, N.F.; Carvalho, C.M.; Paiva, C.N.; Silva Da, A.A. Chronic Trypanosoma cruzi-elicited cardiomyopathy: From the discovery to the proposal of rational therapeutic interventions targeting cell adhesion molecules and chemokine receptors–How to make a dream come true. Mem. Inst. Oswaldo Cruz 2009, 104, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, M.R. Immune-modulating effects of alpha-1 antitrypsin. Biol. Chem. 2014, 395, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Siebers, K.; Fink, B.; Zakrzewicz, A.; Agne, A.; Richter, K.; Konzok, S.; Hecker, A.; Zukunft, S.; Kullmar, M.; Klein, J.; et al. Alpha-1 Antitrypsin Inhibits ATP-Mediated Release of Interleukin-1beta via CD36 and Teceptors. Front. Immunol. 2018, 9, 877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewicz, A.; Richter, K.; Zakrzewicz, D.; Siebers, K.; Damm, J.; Agne, A.; Hecker, A.; Mcintosh, J.M.; Chamulitrat, W.; Krasteva-Christ, G.; et al. SLPI Inhibits ATP-mediated maturation of IL-1beta in human monocytic leukocytes: A Tyer. Front. Immunol. 2019, 10, 664. [Google Scholar] [CrossRef] [Green Version]
- Moreau, T.; Baranger, K.; Dade, S.; Dallet-Choisy, S.; Guyot, N.; Zani, M.L. Multifaceted roles of human elafin and secretory leukocyte proteinase inhibitor (SLPI), two serine protease inhibitors of the chelonianin family. Biochimie 2008, 90, 284–295. [Google Scholar] [CrossRef]
- Ma, G.; Greenwell-Wild, T.; Lei, K.; Jin, W.; Swisher, J.; Hardegen, N.; Wild, C.T.; Wahl, S.M. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 2004, 200, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, P.A.; Alkanani, A.K.; Michels, A.W.; Lewis, E.C.; Shapiro, L.; Dinarello, C.A.; Zipris, D. alpha1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E1418–E1426. [Google Scholar] [CrossRef] [PubMed]
 , increased expression/activity of iPLA2β;
, increased expression/activity of iPLA2β;  , indicates increase in).
, increased expression/activity of iPLA2β; , indicates increase in).
, indicates increase in).
, increased expression/activity of iPLA2β; , indicates increase in). , increased expression/activity of iPLA2β; , indicates increase in; and
, increased expression/activity of iPLA2β; , indicates increase in; and  , indicates decrease in).
, increased expression/activity of iPLA2β; , indicates increase in; and , indicates decrease in).
, indicates decrease in).
, increased expression/activity of iPLA2β; , indicates increase in; and , indicates decrease in).
 , decreased expression/activity of iPLA2β; , increased expression/activity of iPLA2β; , indicates increase in; , indicates decrease in).
, decreased expression/activity of iPLA2β; , increased expression/activity of iPLA2β; , indicates increase in; , indicates decrease in).
, decreased expression/activity of iPLA2β; , increased expression/activity of iPLA2β; , indicates increase in; , indicates decrease in).
, decreased expression/activity of iPLA2β; , increased expression/activity of iPLA2β; , indicates increase in; , indicates decrease in).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, T.D.; Almutairi, A.; Tusing, Y.G.; Lei, X.; Ramanadham, S. The Impact of the Ca2+-Independent Phospholipase A2β (iPLA2β) on Immune Cells. Biomolecules 2021, 11, 577. https://doi.org/10.3390/biom11040577
White TD, Almutairi A, Tusing YG, Lei X, Ramanadham S. The Impact of the Ca2+-Independent Phospholipase A2β (iPLA2β) on Immune Cells. Biomolecules. 2021; 11(4):577. https://doi.org/10.3390/biom11040577
Chicago/Turabian StyleWhite, Tayleur D., Abdulaziz Almutairi, Ying Gai Tusing, Xiaoyong Lei, and Sasanka Ramanadham. 2021. "The Impact of the Ca2+-Independent Phospholipase A2β (iPLA2β) on Immune Cells" Biomolecules 11, no. 4: 577. https://doi.org/10.3390/biom11040577