
Spreading of Beta-Amyloid in Organotypic Mouse Brain Slices and Microglial Elimination and Effects on Cholinergic Neurons

Abstract
:
1. Introduction
1.1. Beta-Amyloid Plaques in Alzheimer’s Disease
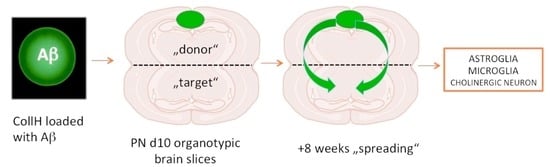
1.2. Spreading in Organotypic Brain Slices
2. Materials and Methods
2.1. Organotypic Brain Slices
2.2. Preparation of Collagen Hydrogels
2.3. Immunohistochemistry
2.4. RNA Isolation and Quantitative TaqMan-PCR
2.5. Western Blots
2.6. Release Experiments
2.7. Data Analysis and Statistics
3. Results
3.1. Culturing of Brain Slices and Viability
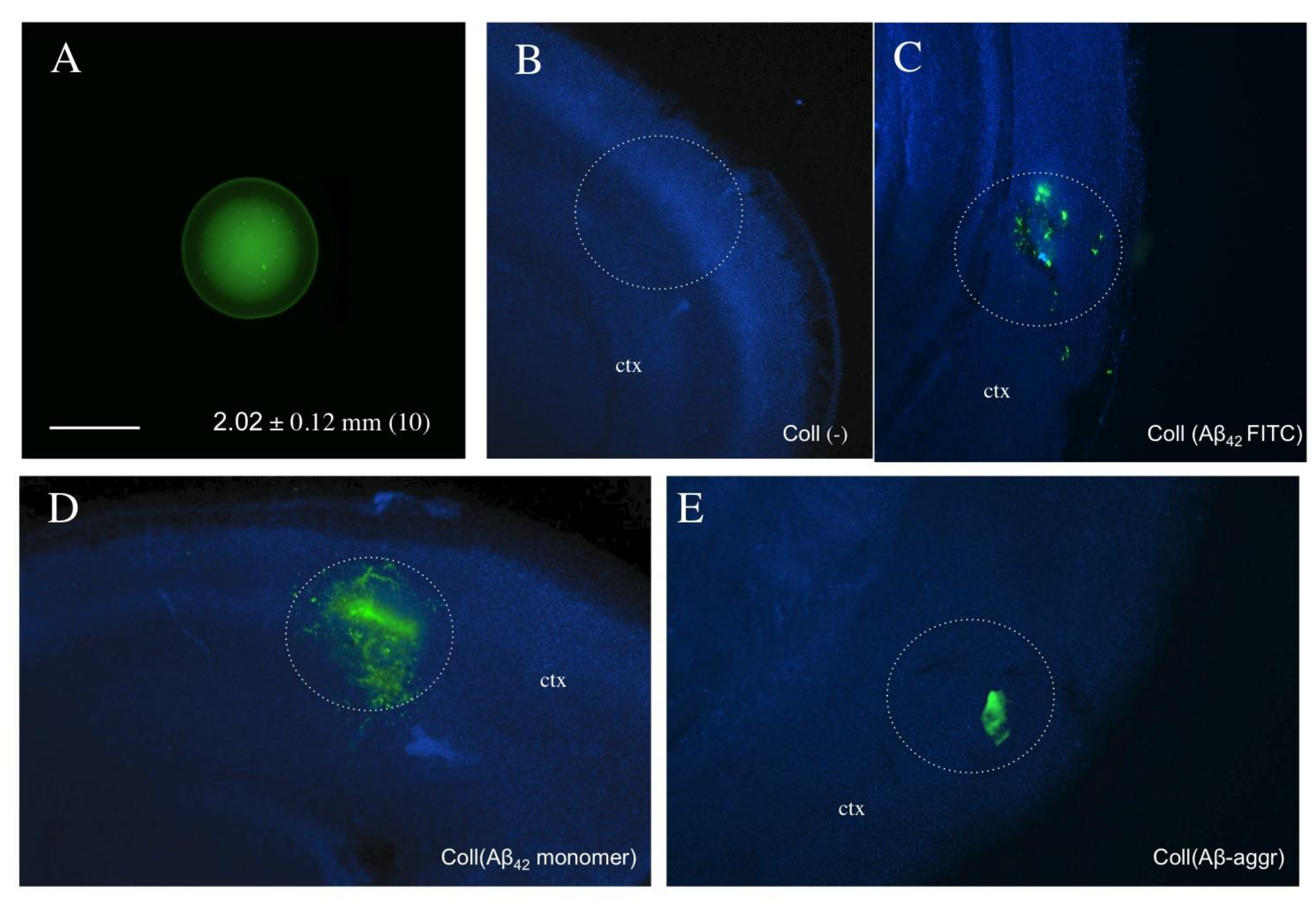
3.2. Loading and Release of Beta-Amyloid from Collagen Hydrogels
3.3. Western Blot of Monomer and Aggregated Aβ
3.4. Beta-Amyloid Spreading in Wildtype Brain Slices
3.5. Beta-Amyloid Spreading in Transgenic APP_SweDI Brain Slices
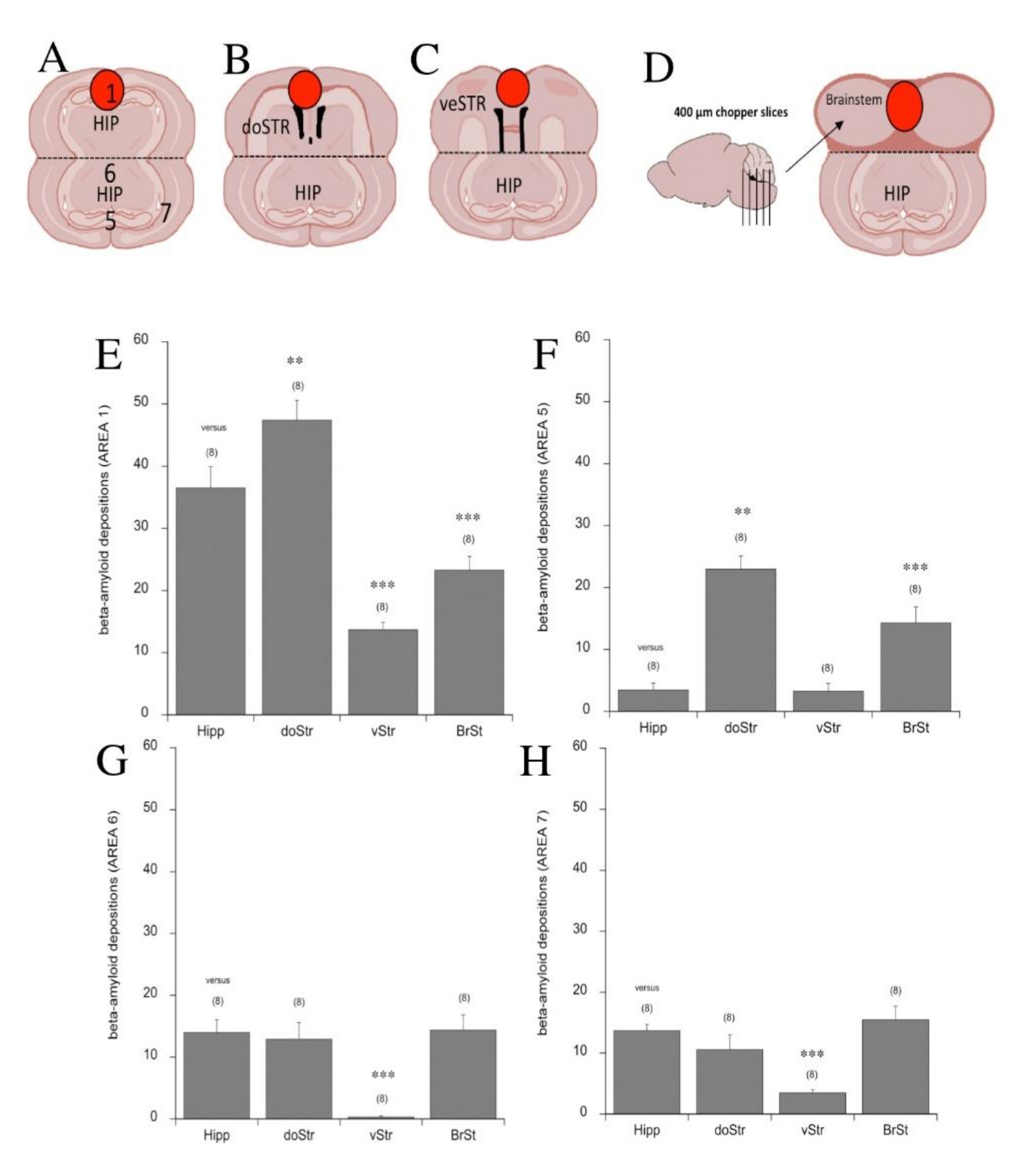
3.6. Beta-Amyloid Spreading in Different Brain Areas
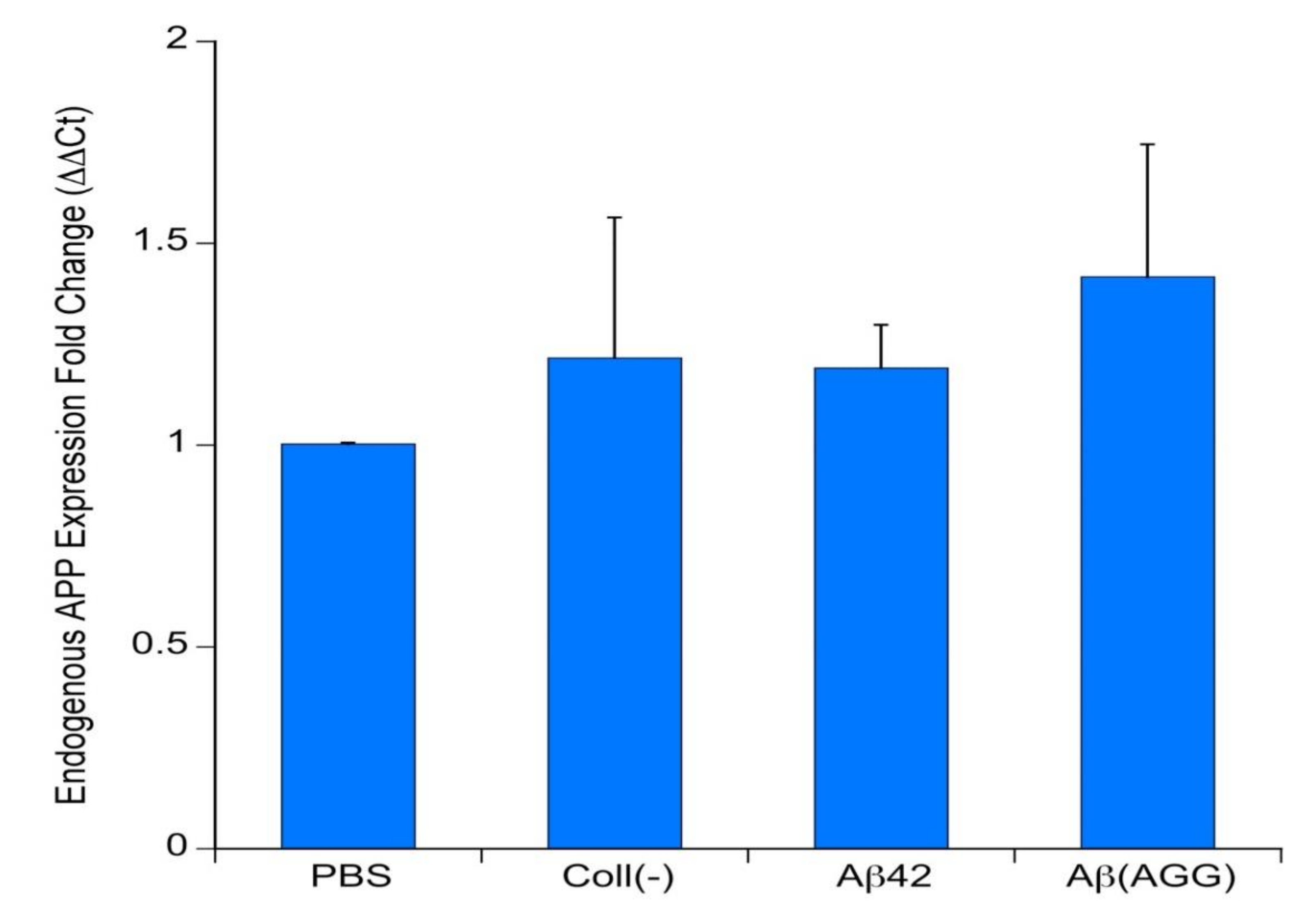
3.7. qRT-PCR of Endogenous APP
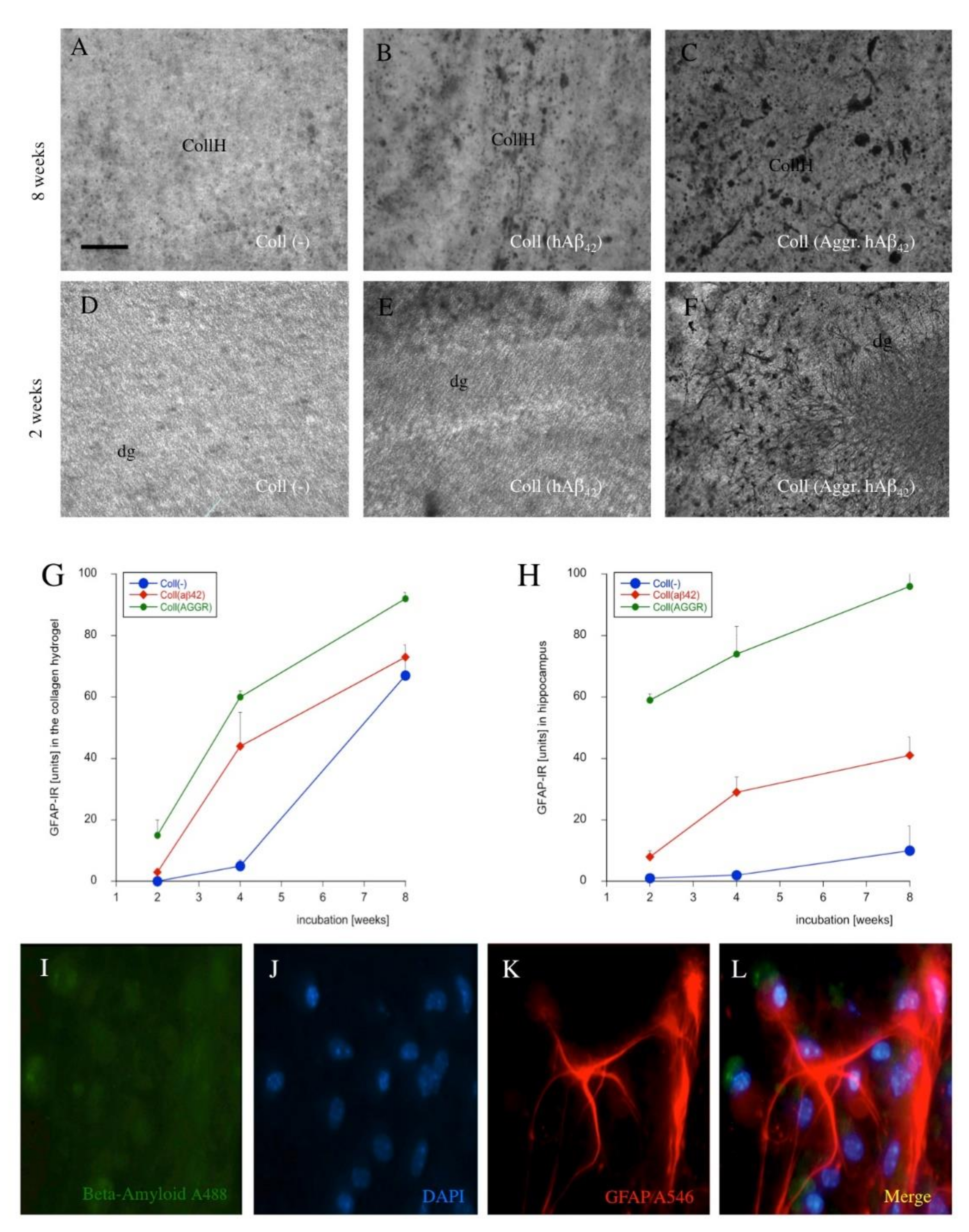
3.8. Astroglial Responses
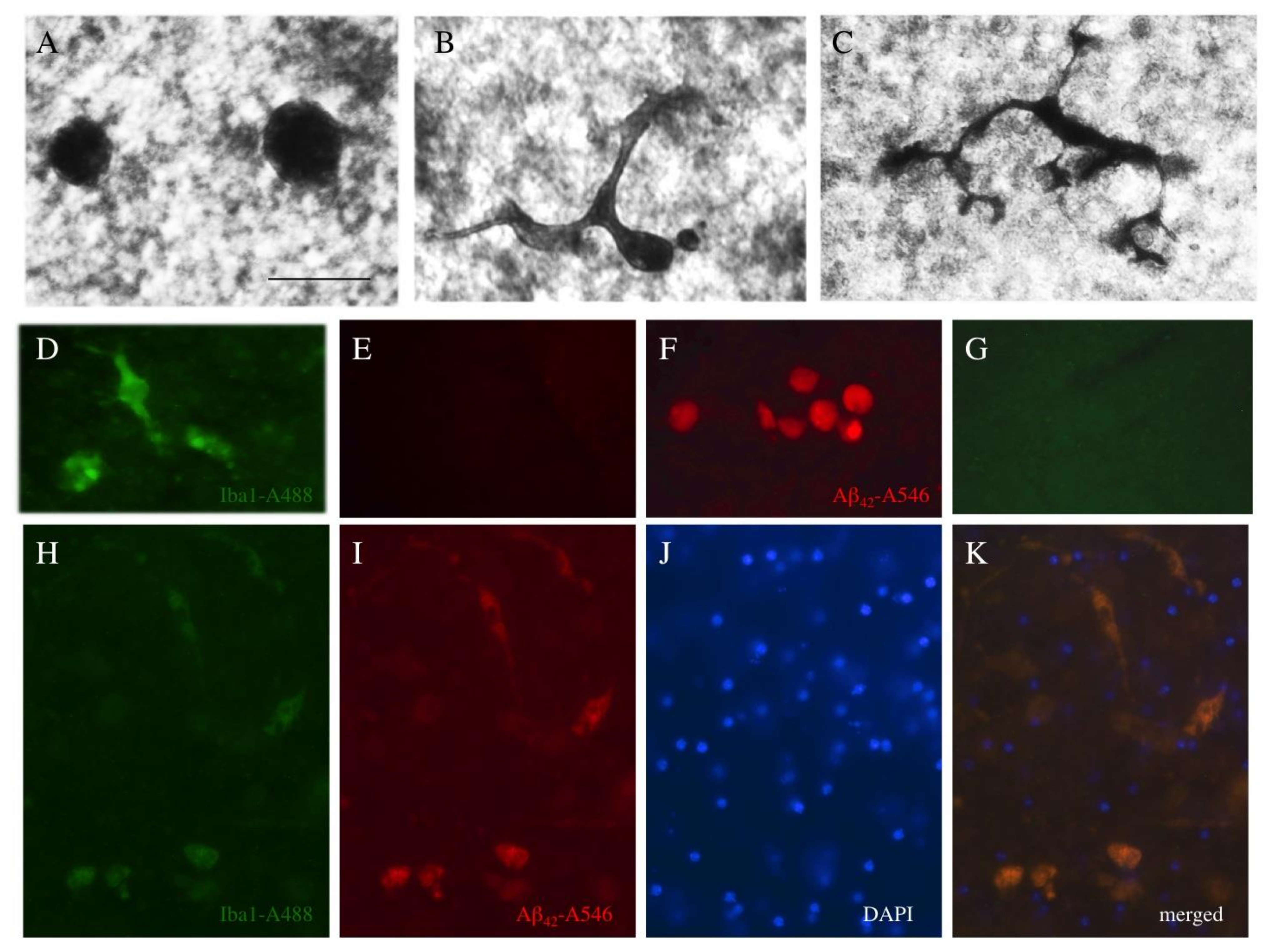
3.9. Microglial Responses
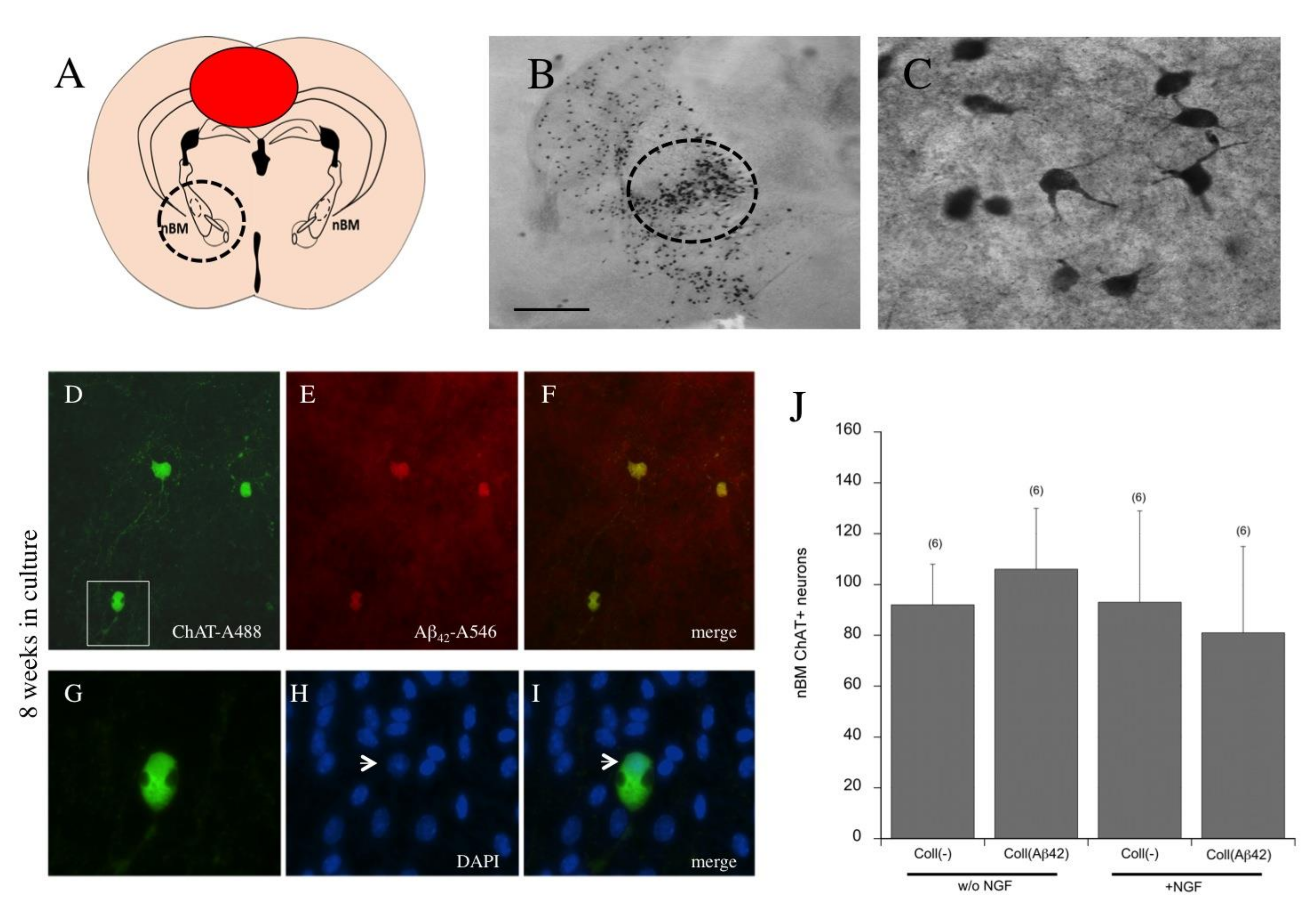
3.10. Effects of Beta-Amyloid Spreading on Cholinergic Neurons
4. Discussion
4.1. Viability of Organotypic Brain Slices to Study Spreading
4.2. Collagen Hydrogels as a Bolus to Apply Substances and Release
4.3. Only Human Beta-Amyloid42 Has Spreading Potential
4.4. Spreading in Transgenic APP Mice
4.5. The Role of Different Brain Areas in Spreading
4.6. Effects on Murine Endogenous APP
4.7. Astroglial Responses
4.8. Microglial Responses
4.9. Effects of Spreading on Cholinergic Neurons
4.10. Spreading Hypothesis
4.11. Mechanism of Spreading
4.12. Translation to Human AD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Attems, J.; Jellinger, K.; Thal, D.R.; van Nostrand, W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2011, 37, 75–93. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Neurodegeneration. Alzheimer’s and Parkinson’s diseases, the prion concept in relation to assembled Aβ, tau and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daschil, N.; Humpel, C. Green-fluorescent protein + Astrocytes Attach to beta-Amyloid Plaques in an Alzheimer Mouse Model and GFP+ astrocytes are Sensitive for Clasmatodendrosis. Front. Aging Neurosci. 2016, 8, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, P.; Seeman, N. Alzheimer’s disease, β-amyloid plaque formation in human brain. Synapse 2011, 65, 1289–1297. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after translation, missorting of tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Alonso, A.C.; Grundke-Iqbal, I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009, 118, 53–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Goedert, M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998, 21, 428–433. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- McAllister, B.B.; Lacoursiere, S.G.; Sutherland, R.J.; Mohajerani, M.H. Intracerebral seeding of amyloid-beta and tau pathology in mice, factors underlying prion-like spreading and comparison with alpha-synuclein. Neurosci. Biobehav. Rev. 2020, 112, 1–27. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Daschil, N.; Kniewallner, K.M.; Obermair, G.J.; Hutter-Paier, B.; Windisch, M.; Marksteiner, J.; Humpel, C. L-Type calcium channel blockers and substance P induce angiogenesis of cortical vessels associated with beta-amyloid plaques in an Alzheimer mouse model. Neurobiol. Aging 2015, 36, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foidl, B.M.; Do-Dinh, P.; Hutter-Schmid, B.; Bliem, H.; Humpel, C. Cholinergic neurodegeneration in an Alzheimer mouse model overexpressing amyloid-precursor protein with the Swedish-Dutch-Iowa mutations. Neurobiol. Learn. Mem. 2016, 136, 86–96. [Google Scholar] [CrossRef]
- Foidl, B.; Humpel, C. Can mouse models mimic sporadic Alzheimer’s disease? Neural Regen. Res. 2019, 15, 401–406. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slices of ADULT transgenic mice, a tool to study Alzheimer´s disease. Curr. Alzheimer Res. 2019, 16, 1–10. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic brain slice cultures-Review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpel, C. Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front. Aging Neurosci. 2015, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Croft, C.L.; Kurbatskaya, K.; Hanger, D.P.; Noble, W. Inhibition of glycogen synthase kinase-3 by BTA-EG4 reduces tau abnormalities in an organotypic brain slice culture model of Alzheimer’s disease. Sci. Rep. 2017, 7, 7434. [Google Scholar] [CrossRef] [Green Version]
- Croft, C.L.; Wade, M.A.; Kurbatskaya, K.; Mastrandreas, P.; Hughes, M.M.; Phillips, E.C.; Pooler, A.M.; Perkinton, M.S.; Hanger, D.P.; Noble, W. Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis. 2017, 8, e2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mewes, A.; Franke, H.; Singer, D. Organotypic brain slice cultures of adult transgenic P301S mice—A model for tauopathy studies. PLoS ONE 2012, 7, e45017. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Noble, W.; Gaynor, K.; Matsuoka, Y. Organotypic slice cultures from transgenic mice as disease model systems. J. Mol. Neurosci. 2002, 19, 317–320. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [Green Version]
- Weis, C.; Marksteiner, J.; Humpel, C. Nerve growth factor and glial cell line-derived neurotrophic factor restore the cholinergic phenotype in organotypic brain slices of the basal nucleus of Meynert. Neuroscience 2001, 102, 129–138. [Google Scholar] [CrossRef]
- Hochstrasser, T.; Hohsfield, L.A.; Sperner-Unterweger, B.; Humpel, C. Beta-amyloid induced effects on cholinergic; serotonergic and dopaminergic neurons is differentially counteracted by anti-inflammatory drugs. J. Neurosci. Res. 2013, 91, 83–94. [Google Scholar]
- Foidl, B.M.; Ucar, B.; Schwarz, A.; Rebel, A.L.; Pandit, A.; Humpel, C. Nerve growth factor released from collagen scaffolds protects axotomized cholinergic neurons of the basal nucleus of Meynert in organotypic brain slices. J. Neurosci. Methods 2018, 295, 77–86. [Google Scholar] [CrossRef]
- Marksteiner, J.; Humpel, C. Beta-amyloid expression; release and extracellular deposition in aged rat brain slices. Mol. Psychiatry 2008, 13, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.A.; Narrow, W.C.; Federoff, H.J.; Bowersm, W.J. An improved method for generating consistent soluble amyloid-beta oligomer preparations for in vitro neurotoxicity studies. J. Neurosci. Methods 2010, 190, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foidl, M.; Humpel, C. Differential hyperphosphorylation of tau-S199; -T231 and -S396 in organotypic brain slices of Alzheimer mice. A model to study early tau hyperphosphorylation using Okadaic acid. Front. Ageing Neurosci. 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daschil, N.; Geisler, S.; Obermair, G.J.; Humpel, C. Short and long-term treatment of mouse cortical primary astrocytes with β-amyloid differentially regulate the mRNA expression of L-type calcium channels. Pharmacology 2014, 93, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineau, H.; Sim, V. POSCAbilities, The Application of the Prion Organotypic Slice Culture Assay to Neurodegenerative Disease, Research. Biomolecules 2020, 10, 1079. [Google Scholar] [CrossRef] [PubMed]
- Ucar, B.; Humpel, C. Collagen for brain repair, therapeutic perspectives-a mini-review. Neural Regen. Res. 2018, 13, 595–598, Review. [Google Scholar] [CrossRef]
- Ucar, B.; Kajtez, J.; Foidl, B.M.; Eigel, D.; Werner, C.; Long, K.R.; Emnéus, J.; Bizeau, J.; Lomora, M.; Pandit, A.; et al. Biomaterial based strategies to reconstruct the nigrostriatal pathway in organotypic slice co-cultures. Acta Biomater. 2021, 23. [Google Scholar] [CrossRef]
- Ucar, B.; Yusufogullari, S.; Humpel, C. Collagen hydrogels loaded with fibroblast growth factor-2 as a bridge to repair brain vessels in organotypic brain slices. Exp. Brain Res. 2020, 238, 2521–2529. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; de Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of beta-amyloid: Where the Alzheimer’s disease protein meets physiology. Rev. Neurosci. 2010, 21, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Sepulcre, J.; Grothe, M.J.; D’Oleire Uquillas, F.; Ortiz-Terán, L.; Diez, I.; Yang, H.S.; Jacobs, H.I.L.; Hanseeuw, B.J.; Li, Q.; El-Fakhri, G.; et al. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nat. Med. 2018, 24, 1910–1918. [Google Scholar] [CrossRef]
- Delacourte, A.; David, J.P.; Sergeant, N.; Buée, L.; Wattez, A.; Vermersch, P.; Ghozali, F.; Fallet-Bianco, C.; Pasquier, F.; Lebert, F.; et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999, 52, 1158–1165. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Dudvarski Stankovic, N.; Teodorczyk, M.; Ploen, R.; Zipp, F.; Schmidt, M.H.H. Microglia-blood vessel interactions, a double-edged sword in brain pathologies. Acta Neuropathol. 2016, 131, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.D.; Tsai, C.A.; Rubin, A.J.; James, M.L.; Andreasson, K.I. Microglial malfunction, the third rail in the development of Alzheimer’s disease. Trends Neurosci. 2015, 38, 621–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis, Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Färber, K.; Kettenmann, H. Physiology of microglial cells. Brain Res. Rev. 2004, 48, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Hellwig, S.; Masuch, A.; Nestel, S.; Katzmarski, N.; Meyer-Luehmann, M.; Biber, K. Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-β plaque formation in organotypic hippocampal slice cultures. Sci. Rep. 2015, 5, 14624. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Brazil, M.I.; Soe, T.T.; Maxfield, F.R. Uptake; degradation and release of fibrillar and soluble forms of Alzheimer’s amyloid beta-peptide by microglial cells. J. Biol. Chem. 1999, 274, 32301–32308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Vidovic, N.; Biber, K.; Dolga, A.; Culmsee, C.; Dodel, R. The neuroprotective role of microglial cells against amyloid beta-mediated toxicity in organotypic hippocampal slice cultures. Brain Pathol. 2020, 30, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Gandy, S.; Heppner, F.L. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsay, H.J.; Huang, Y.C.; Huang, F.L.; Chen, C.P.; Tsai, Y.C.; Wang, Y.H.; Wu, M.F.; Chiang, F.Y.; Shiao, Y.J. Amyloid beta peptide-mediated neurotoxicity is attenuated by the proliferating microglia more potently than by the quiescent phenotype. J Biomed. Sci. 2013, 20, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Mezias, C.; Raj, A. Analysis of Amyloid-β Pathology Spread in Mouse Models Suggests Spread Is Driven by Spatial Proximity, Not Connectivity. Front. Neurol. 2017, 8, 653. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for Seeding of β-Amyloid by Intracerebral Infusion of Alzheimer Brain Extracts in β-Amyloid Precursor Protein-Transgenic Mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Bravo-Alegria, J.; Duran-Aniotz, C.; Soto, C. Titration of biologically active amyloid–β seeds in a transgenic mouse model of Alzheimer’s disease. Sci. Rep. 2015, 5, 9349. [Google Scholar] [CrossRef]
- Pignataro, A.; Middei, S. Trans-Synaptic Spread of Amyloid- β in Alzheimer’s Disease, Paths to β—Amyloidosis. Neural Plast. 2017, 2017, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Evidence for trans-synaptic and exo-synaptic tau propagation in Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 665. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 2016, 17, 251–260. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Borroto-Escuela, D.O. Volume transmission and receptor-receptor interactions in heteroreceptor complexes, understanding the role of new concepts for brain communication. Neural Regen. Res. 2016, 11, 1220–1223. [Google Scholar] [CrossRef]
- Humpel, C. Intranasal delivery of collagen-loaded neprilysin clears beta-amyloid plaques in a transgenic Alzheimer mouse model. Front. Aging Neurosci. 2021. Revision. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Area 1 | Area 2 | Area 3 | Area 4 | Area 5 | Area 6 | Area 7 |
|---|---|---|---|---|---|---|---|
| PBS | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns |
| CollH (–) | 0 ± 0 (20) vs | 0 ± 0 (20) vs | 0 ± 0 (20) vs | 0 ± 0 (19) vs | 0 ± 0 (20) vs | 0 ± 0 (20) vs | 0 ± 0 (21) vs |
| CollH (hAβ40) | 0.4 ± 0.4 (5) ns | 0.4 ± 0.4 (5) ns | 0 ± 0 (5) ns | 0 ± 0 (5) ns | 0 ± 0 (5) ns | 0 ± 0 (5) ns | 0 ± 0 (5) ns |
| CollH (hAβ42) | 54.7 ± 9.7 (24) *** | 26.2 ± 7.3 (24) *** | 9.8 ± 2.4 (24) *** | 3.8 ± 1 (24) ** | 1.6 ± 0.5 (22) * | 5.5 ± 1.6 (22) * | 2.1 ± 0.8 (24) ** |
| CollH (AGG hAβ42) | 60 ± 8.2 (18) *** | 32.3 ± 9.5 (18) ** | 7.7 ± 1.7 (17) ** | 4.8 ± 1.1 (18) ns | 1.3 ± 0.6 (19) * | 9.2 ± 2.6 (19) *** | 1.6 ± 0.4 (19) ns |
| hAβ42 (w/o CollH) | 0.3 ± 0.3 (6) ns | 4.7 ± 4.7 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0.2 ± 0.2 (6) ns | 0 ± 0 (3) ns |
| CollH (pyrG-Aβ) | 1 ± 1 (3) ns | 9.3 ± 6.4 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 1.3 ± 0.7 (3) ns | 0 ± 0 (3) ns |
| CollH (Aβ25-35) | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns | 0 ± 0 (3) ns |
| CollH (hAβ42Rev) | 14.7 ± 5.0 (6) ns | 13.8 ± 3.5 (6) ns | 16.6 ± 1.6 (6) *** | 16.7 ± 1.0 (6) *** | 0.5 ± 0.2 (6) ns | 1.0 ± 0.4 (6) ns | 0 ± 0 (6) ns |
| CollH (mAβ42) | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns | 0 ± 0 (6) ns |
| Group | Area 1 | Area 3 | Area 5 | Area 7 | |
|---|---|---|---|---|---|
| PBS | Round | 9.4 ± 1.7 (6) ** | 7.4 ± 0.5 (6) ** | 4.6 ± 0.5 (6) *** | 3.6 ± 0.6 (6) *** |
| Ramified | 1.6 ± 0.3 (6) ns | 2.3 ± 0.5 (6) ns | 1.4 ± 0.3 (6) ns | 0.2 ± 0.2 (6) ** | |
| Total | 11 ± 2.0 (6) ** | 9.7 ± 1.0 (6) ns | 6.0 ± 0.8 (6) ns | 3.8 ± 0.8 (6) ns | |
| CollH(-) | Round | 16.2 ± 0.9 (6) vs | 17.6 ± 0.6 (6) vs | 14.6 ± 1.4 (6) vs | 12.8 ± 1.2 (6) vs |
| Ramified | 4.1 ± 1.4 (6) vs | 2.3 ± 0.8 (6) vs | 3.5 ± 0.9 (6) vs | 3 ± 0.4 (6) vs | |
| Total | 20.3 ± 2.3 (6) vs | 19.9 ± 1.4 (6) vs | 18.1 ± 2.7 (6) vs | 15.8 ± 1.6 (6) vs | |
| CollH(Aβ42) | Round | 25.3 ± 1.1 (6) *** | 24.6 ± 1.2 (6) *** | 15.4 ± 0.9 (6) ns | 12.6 ± 2.3 (6) ns |
| Ramified | 10.2 ± 1.3 (6) *** | 5.8 ± 1.2 (6) ns | 4.2 ± 1.2 (6) ns | 3.6 ± 1.5 (6) ns | |
| Total | 35.5 ± 2.4 (6) ** | 30.4 ± 2.4 (6) ns | 19.6 ± 2.1 (6) ns | 16.2 ± 3.8 (6) ns | |
| CollH(AGG Aβ42) | Round | 22.8 ± 1.1 (6) *** | 25.6 ± 1.8 (6) *** | 16 ± 1.4 (6) ns | 13.4 ± 1.8 (6) ns |
| Ramified | 8.5 ± 0.9 (6) ns | 12.4 ± 3.1 (6) *** | 1.5 ± 0.6 (6) ns | 2.4 ± 0.5 (6) ns | |
| Total | 31.3 ± 2 (6) ** | 38 ± 4.9. (6) ** | 17.5 ± 2 (6) ns | 15.8 ± 2.3 (6) ns | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moelgg, K.; Jummun, F.; Humpel, C. Spreading of Beta-Amyloid in Organotypic Mouse Brain Slices and Microglial Elimination and Effects on Cholinergic Neurons. Biomolecules 2021, 11, 434. https://doi.org/10.3390/biom11030434
Moelgg K, Jummun F, Humpel C. Spreading of Beta-Amyloid in Organotypic Mouse Brain Slices and Microglial Elimination and Effects on Cholinergic Neurons. Biomolecules. 2021; 11(3):434. https://doi.org/10.3390/biom11030434
Chicago/Turabian StyleMoelgg, Kurt, Faryal Jummun, and Christian Humpel. 2021. "Spreading of Beta-Amyloid in Organotypic Mouse Brain Slices and Microglial Elimination and Effects on Cholinergic Neurons" Biomolecules 11, no. 3: 434. https://doi.org/10.3390/biom11030434