
Comprehensive Characterization of the Coding and Non-Coding Single Nucleotide Polymorphisms in the Tumor Protein p63 (TP63) Gene Using In Silico Tools

 ,
,  , and
, and
Abstract
:1. Introduction
- Determine the consequences of various nsSNPs in the human TP63 gene on the TP63 protein using different in silico tools.
- Evaluate the effect of nsSNPs on the binding affinity of the TP63 protein with its ligands (DNA) by molecular docking to confirm the consequences.
- Simulate interactions of DNA and TP63 protein with molecular dynamics simulations to validate the effect on protein function caused by high impact nsSNPs.
2. Methods and Materials
2.1. SNP Data Retrieval
2.2. Sequence Investigation (Functional Consequence Analysis of nsSNPs)
2.3. Structural Modeling
2.4. Molecular Docking
2.5. Molecular Dynamics Simulation
2.6. Analysis of the Functional Consequences of Non-Coding SNPs
3. Results
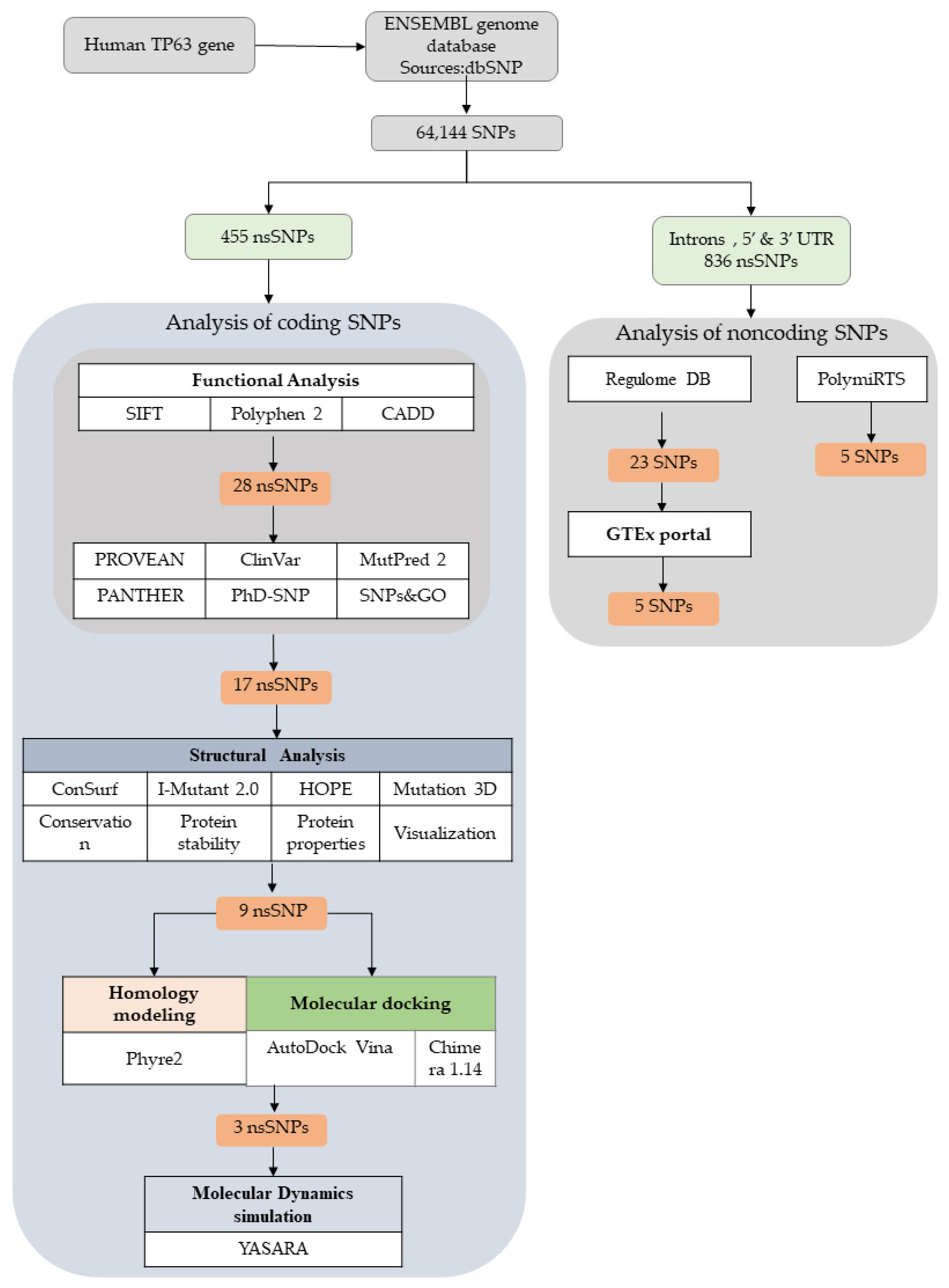
3.1. TP63 SNP Data Retrieval
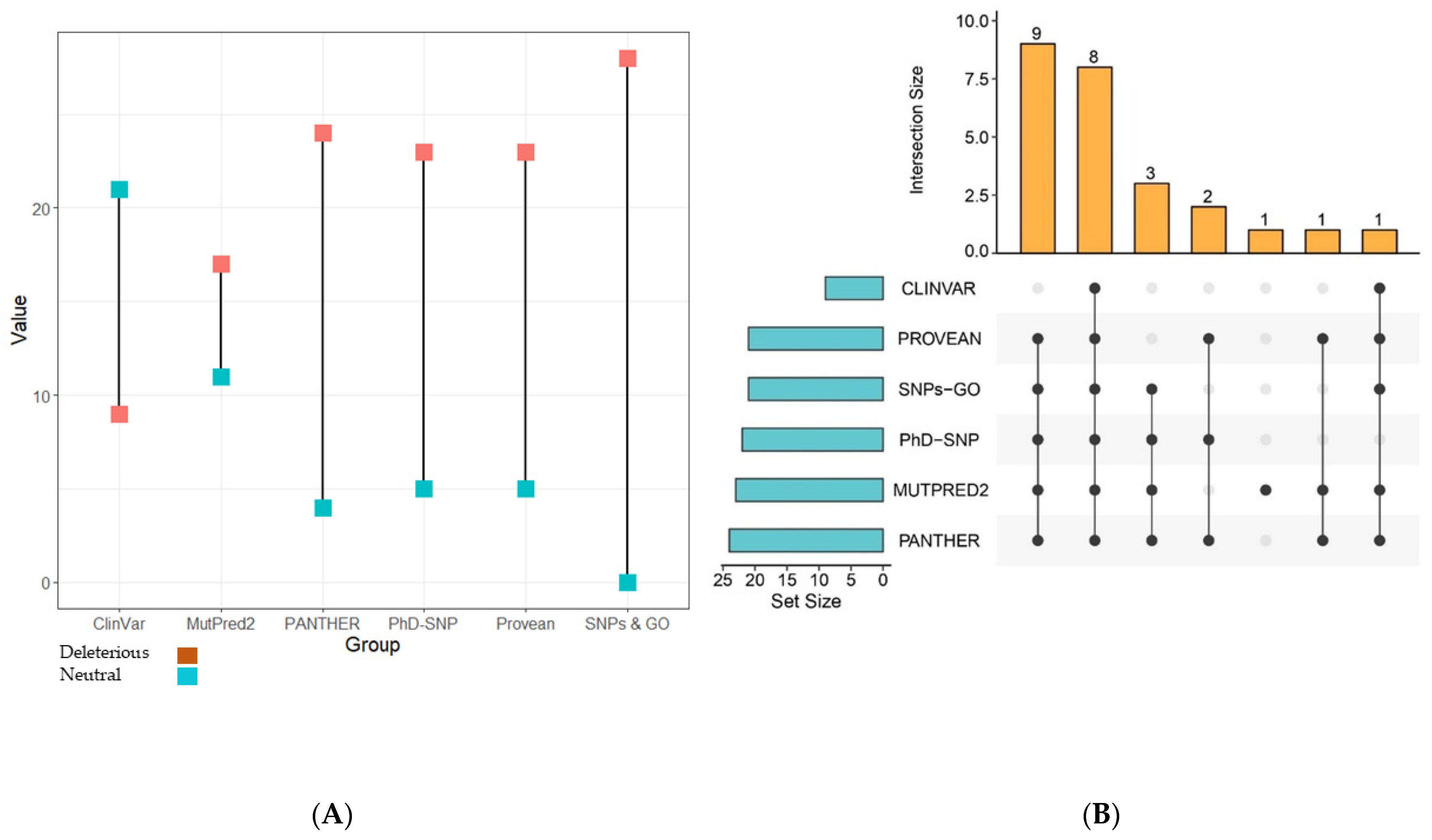
3.2. Prediction of Functionally Important nsSNPs in the TP63 Gene
3.3. Domain Identification for nsSNPs
3.4. Structural Analysis
3.4.1. I-Mutant 2.0 Analysis
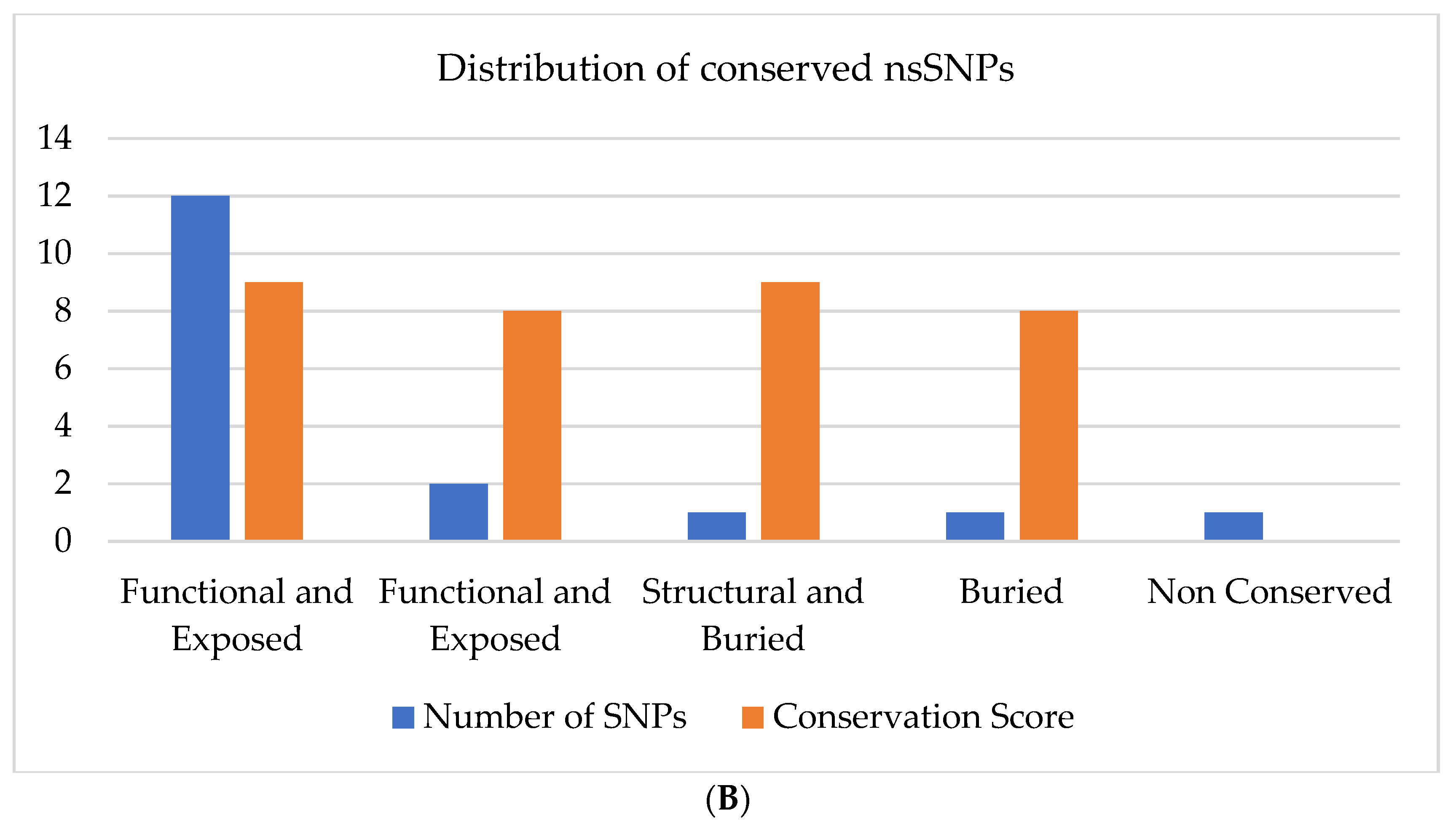
3.4.2. Effect of nsSNPs on Evolutionary Conservation of TP63 Protein Using Consurf
3.4.3. Mutation 3D Analysis
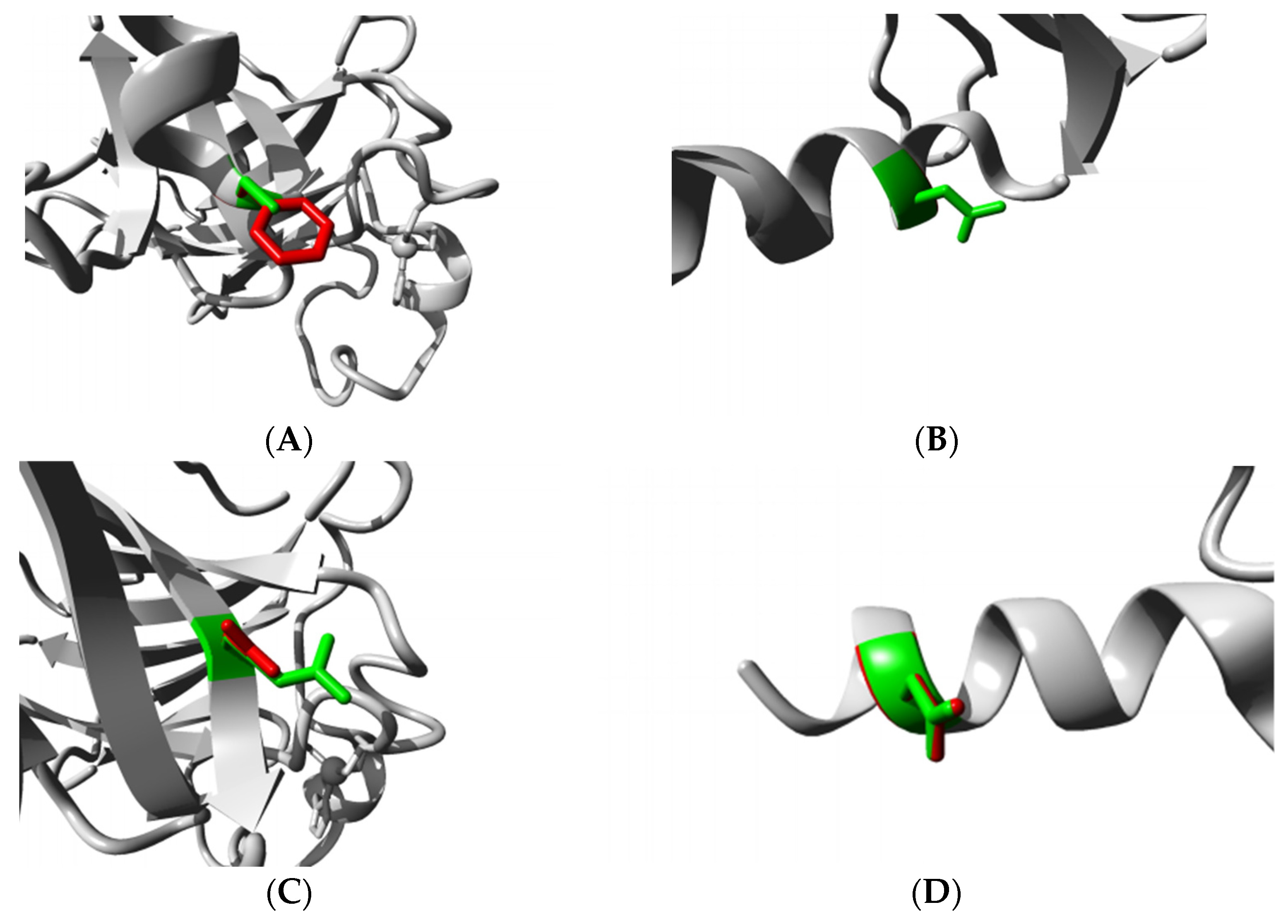
3.4.4. HOPE Analysis
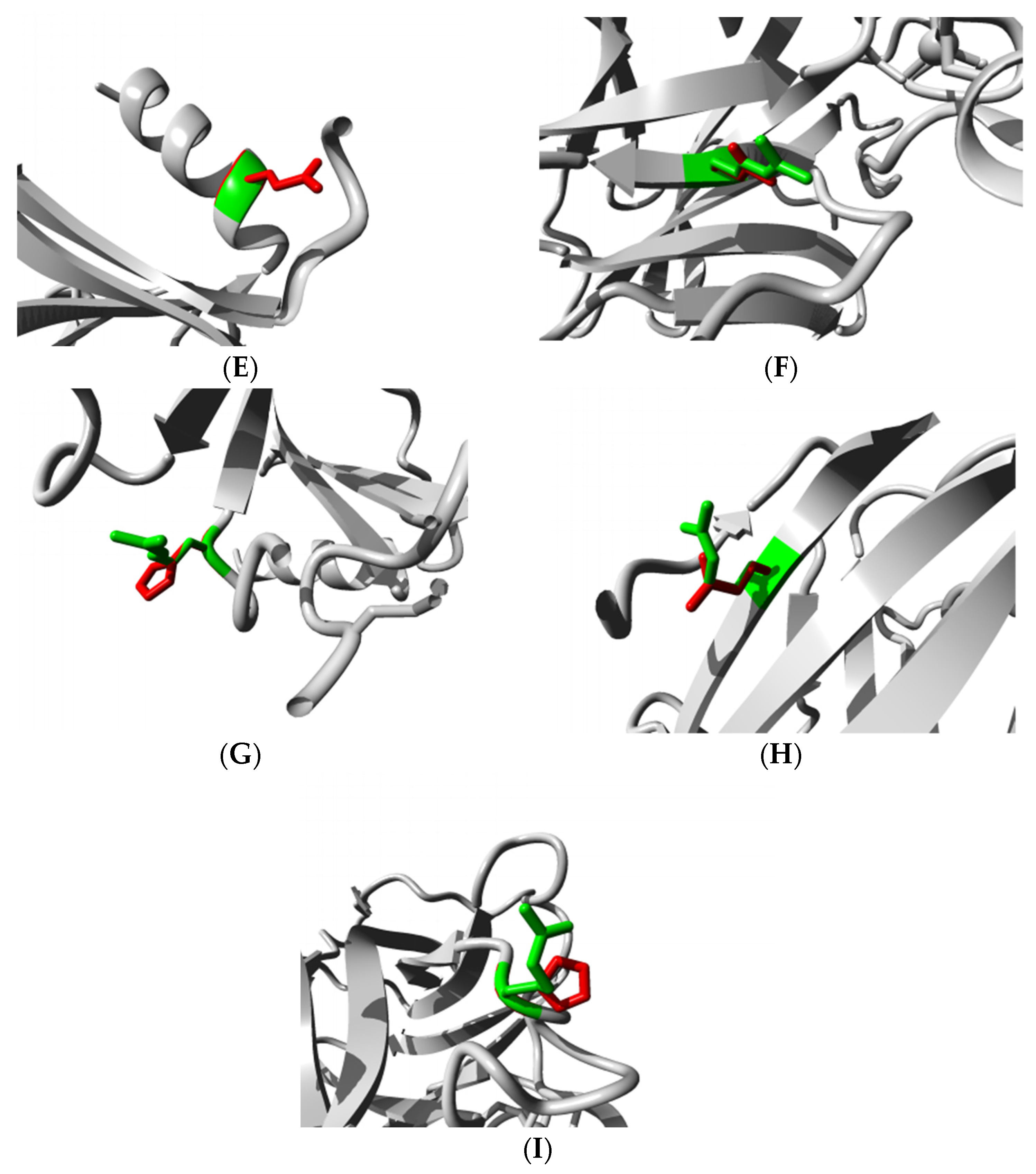
3.5. Structural Effect Analysis of nsSNPs
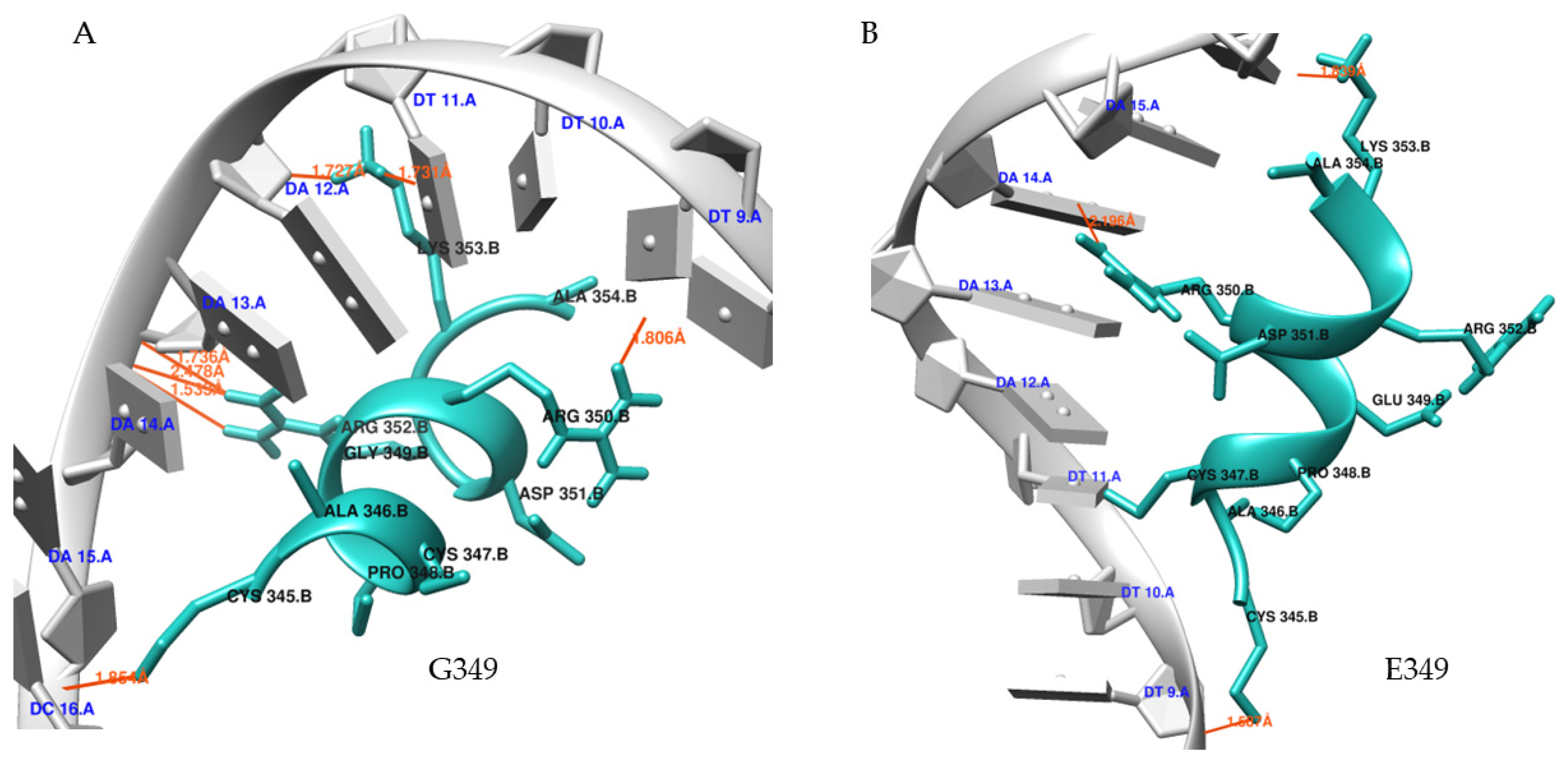
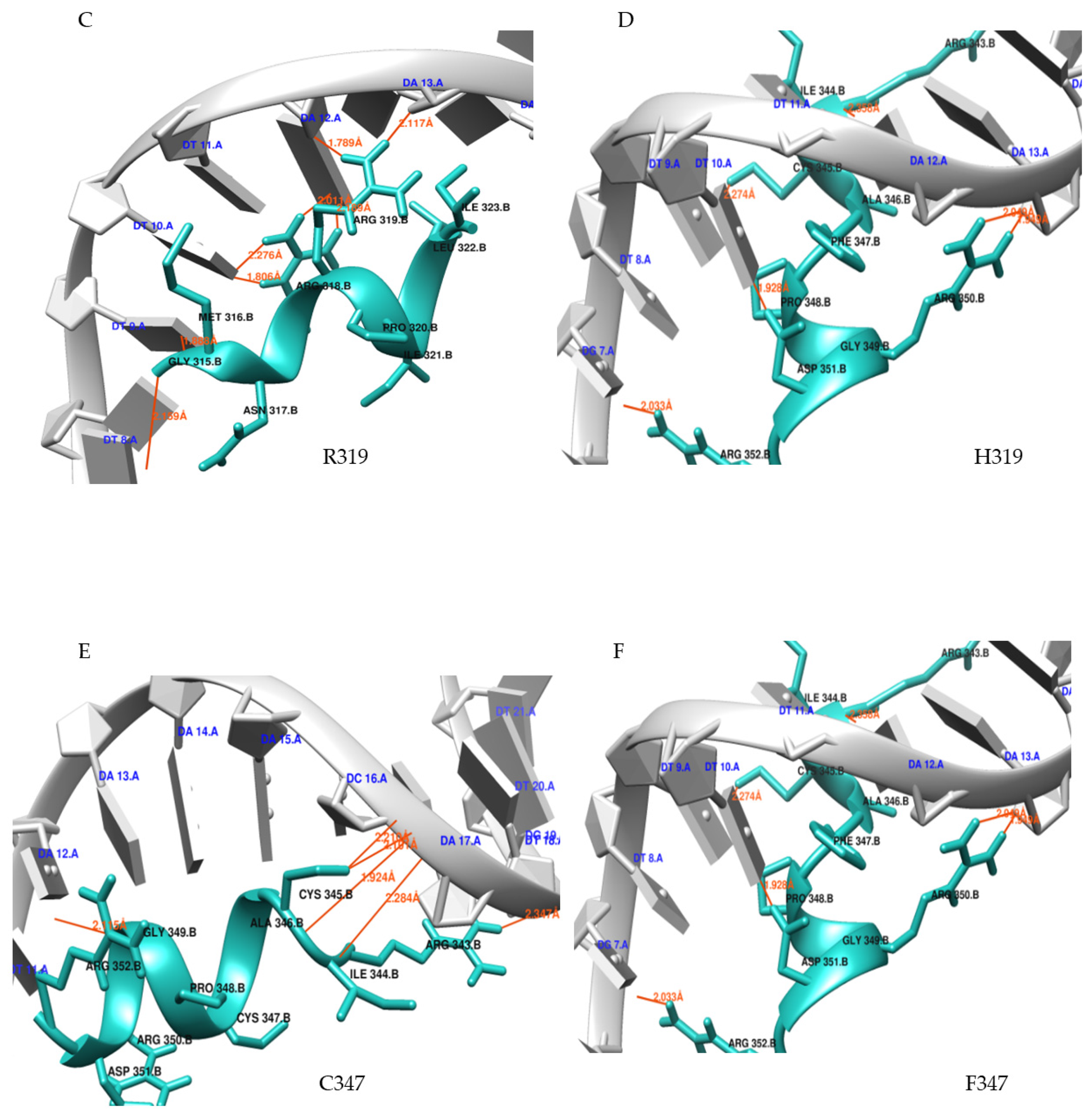
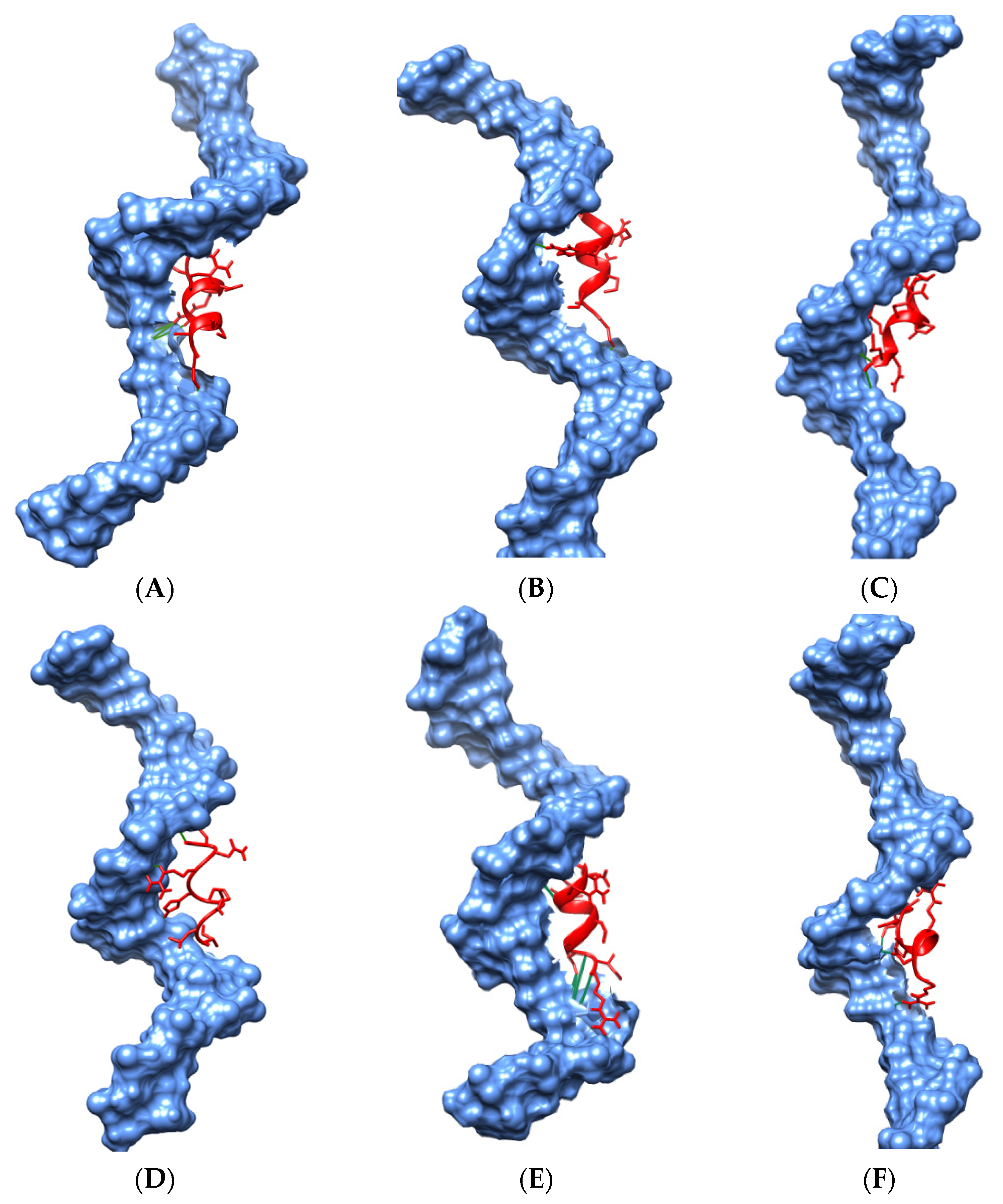
Molecular Docking
3.6. Molecular Dynamics (MD) Simulations
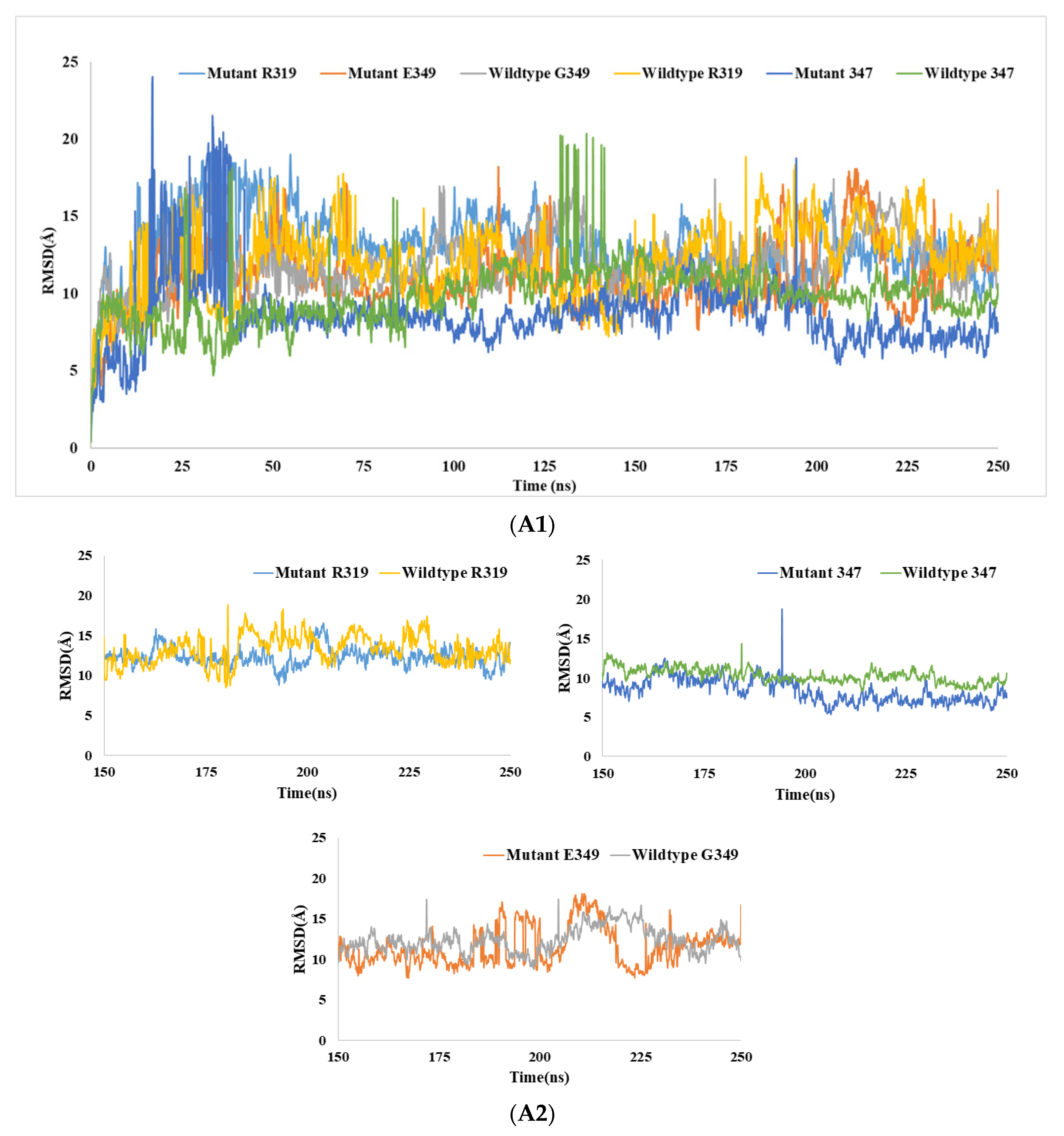
3.6.1. RMSD Analysis
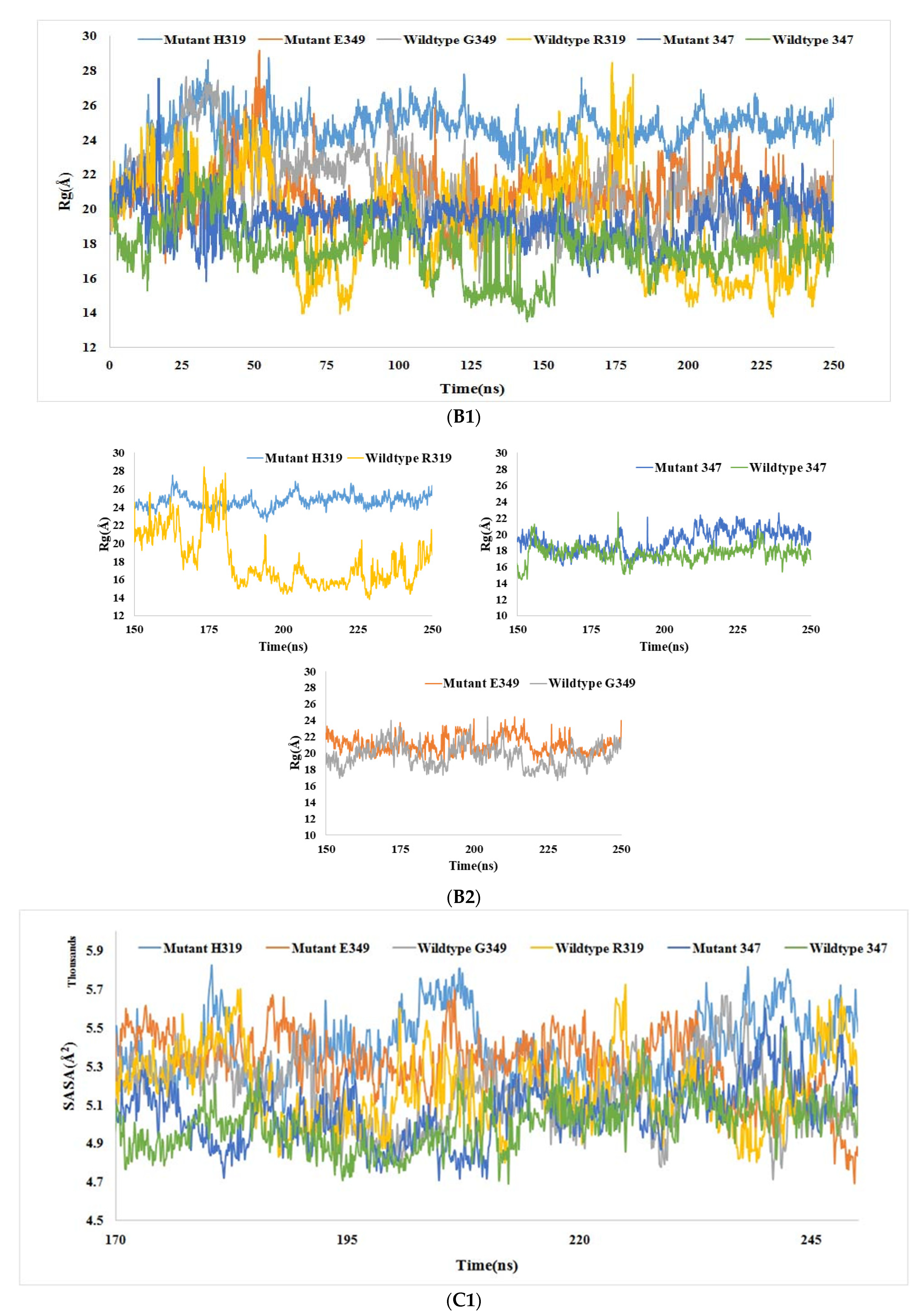
3.6.2. Rg Analysis
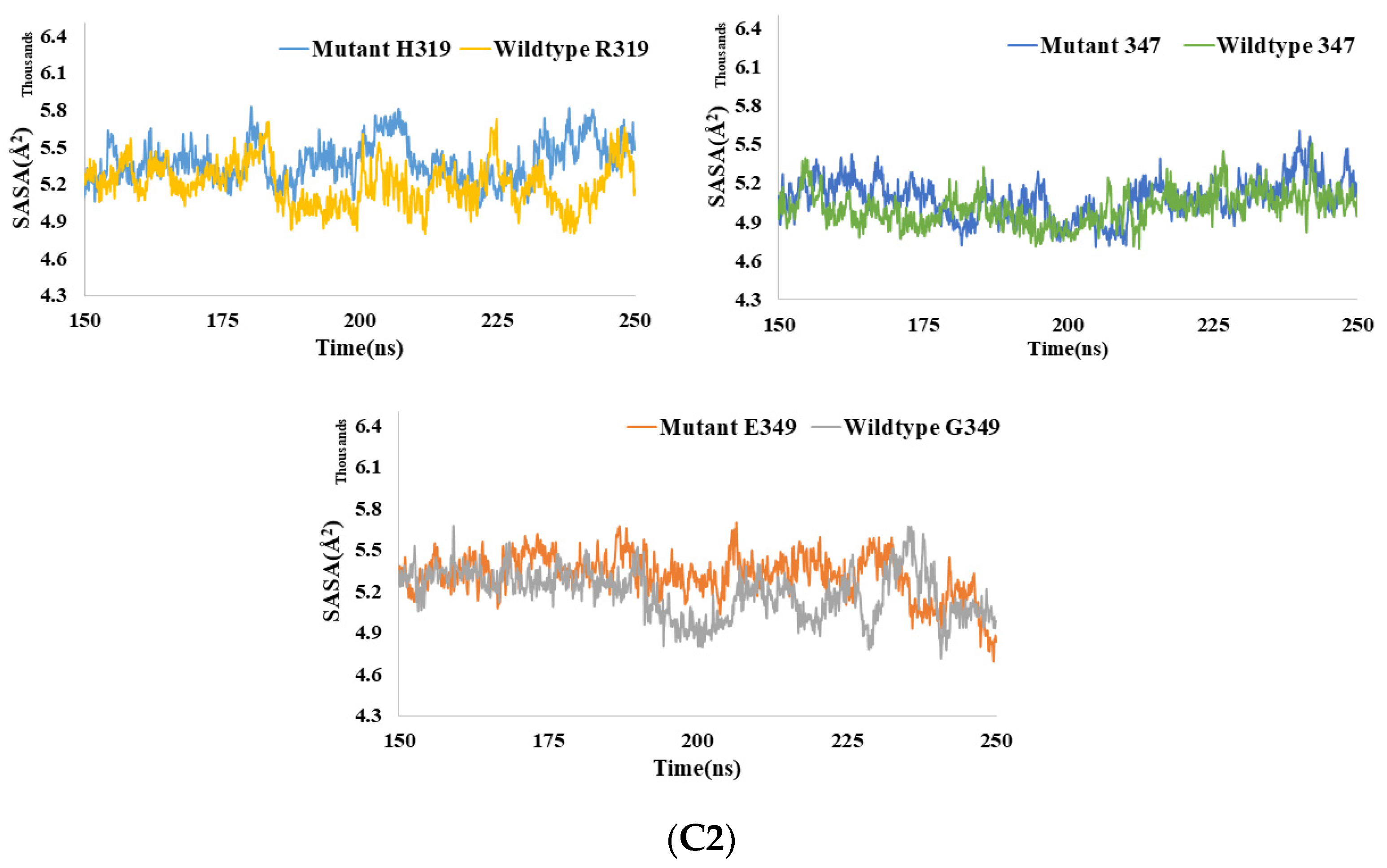
3.6.3. SASA Analysis
3.7. Analysis of Non-Coding SNPs
3.7.1. RegulomeDB Analysis
3.7.2. Finding eQTLs Using GTEX Analysis
3.7.3. PolymiRTS Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Monti, P.; Ciribilli, Y.; Foggetti, G.; Menichini, P.; Bisio, A.; Cappato, S.; Inga, A.; Divizia, M.T.; Lerone, M.; Bocciardi, R.; et al. P63 modulates the expression of the WDFY2 gene which is implicated in cancer regulation and limb development. Biosci. Rep. 2019, 39, BSR20192114. [Google Scholar] [CrossRef] [PubMed]
- Ghioni, P.; Bolognese, F.; Duijf, P.H.G.; van Bokhoven, H.; Mantovani, R.; Guerrini, L. Complex Transcriptional Effects of p63 Isoforms: Identification of Novel Activation and Repression Domains. Mol. Cell. Biol. 2002, 22, 8659–8668. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- McDade, S.S.; Patel, D.; Moran, M.; Campbell, J.; Fenwick, K.; Kozarewa, I.; Orr, N.J.; Lord, C.J.; Ashworth, A.A.; McCance, D.J. Genome-wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res. 2014, 42, 6270–6285. [Google Scholar] [CrossRef]
- Amiel, J.; Bougeard, G.; Francannet, C.; Raclin, V.; Munnich, A.; Lyonnet, S.; Frebourg, T. TP63 gene mutation in ADULT syndrome. Eur. J. Hum. Genet. 2001, 9, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Kiemeney, L.A.; Thorlacius, S.; Sulem, P.; Geller, F.; Aben, K.K.H.; Stacey, S.N.; Gudmundsson, J.; Jakobsdottir, M.; Bergthorsson, J.T.; Sigurdsson, A.; et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat. Genet. 2008, 40, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Vanbokhoven, H.; Melino, G.; Candi, E.; Declercq, W. P63, a story of mice and men. J. Investig. Dermatol. 2011, 131, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Miki, D.; Kubo, M.; Takahashi, A.; Yoon, K.A.; Kim, J.; Lee, G.K.; Zo, J.I.; Lee, J.S.; Hosono, N.; Morizono, T.; et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet. 2010, 42, 893–896. [Google Scholar] [CrossRef]
- Duijf, P.H.G.; Vanmolkot, K.R.J.; Propping, P.; Friedl, W.; Krieger, E.; McKeon, F.; Dötsch, V.; Brunner, H.G.; van Bokhoven, H. Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum. Mol. Genet. 2002, 11, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; McMenamin, M.G.; Miura, K.; Harris, C.C. Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res. 1999, 59, 4165–4169. [Google Scholar]
- Gassoum, A.; Abdelraheem, N.E.; Elsadig, N. Comprehensive Analysis of rsSNPs Associated with Hypertension Using In-Silico Bioinformatics Tools. OALib 2016, 3, 1–24. [Google Scholar] [CrossRef]
- Desai, M.; Chauhan, J. In silico analysis of nsSNPs in human methyl CpG binding protein 2. Meta Gene 2016, 10, 1–7. [Google Scholar] [CrossRef]
- Hossain, S.; Hosen, M.I. Dissecting the structural and functional impact of SNPs located in the spinal muscular atrophy associated gene SMN1 using in silico analysis. Gene Rep. 2019, 15, 100388. [Google Scholar] [CrossRef]
- Cargill, M.; Altshuler, D.; Ireland, J.; Sklar, P.; Ardlie, K.; Patil, N.; Shaw, N.; Lane, C.R.; Lim, E.P.; Kalyanaraman, N.; et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999, 22, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Wigner, P.; Strycharz, J.; Watala, C.; Swiderska, E.; Synowiec, E.; Galecki, P.; Talarowska, M.; Szemraj, J.; Su, K.P.; et al. Single-nucleotide polymorphisms of uracil-processing genes affect the occurrence and the onset of recurrent depressive disorder. PeerJ 2018, 2018, e5116. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Chauhan, J.B. Computational analysis for the determination of deleterious nsSNPs in human MTHFD1 gene. Comput. Biol. Chem. 2017, 70, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Divanshu, G.; Lekshmi, M.; Shanthi, V. In silico studies of deleterious non-synonymous single nucleotide polymorphisms (nsSNPs) of NRL gene. Netw. Model. Anal. Health Inform. Bioinform. 2014, 3, 1–7. [Google Scholar] [CrossRef]
- Nimir, M.; Abdelrahim, M.; Abdelrahim, M.; Abdalla, M.; Ahmed, W.e.; Abdullah, M.; Hamid, M.M.A. In silico analysis of single nucleotide polymorphisms (SNPs) in human FOXC2 gene. F1000Research 2017, 6, 243. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, C.; Wang, Z.; Lv, H.; Li, X. In silico analysis of non-synonymous single nucleotide polymorphisms (nsSNPs) in the human GJA3 gene associated with congenital cataract. BMC Mol. Cell Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Badgujar, N.V.; Tarapara, B.V.; Shah, F.D. Computational analysis of high-risk SNPs in human CHK2 gene responsible for hereditary breast cancer: A functional and structural impact. PLoS ONE 2019, 14, e0220711. [Google Scholar] [CrossRef]
- Islam, M.J.; Khan, A.M.; Parves, M.R.; Hossain, M.N.; Halim, M.A. Prediction of Deleterious Non-synonymous SNPs of Human STK11 Gene by Combining Algorithms, Molecular Docking, and Molecular Dynamics Simulation. Sci. Rep. 2019, 9, 16426. [Google Scholar] [CrossRef] [PubMed]
- Doss, C.G.P.; Chakraborty, C.; Rajith, B.; Nagasundaram, N. In silico discrimination of nsSNPs in hTERT gene by means of local DNA sequence context and regularity. J. Mol. Model. 2013, 19, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Abdul Samad, F.; Suliman, B.A.; Basha, S.H.; Manivasagam, T.; Essa, M.M. A comprehensive In Silico analysis on the structural and functional impact of SNPs in the congenital heart defects associated with NKX2-5 gene—A molecular dynamic simulation approach. PLoS ONE 2016, 11, e0153999. [Google Scholar] [CrossRef]
- Havranek, B.; Islam, S.M. Prediction and evaluation of deleterious and disease causing non-synonymous SNPs (nsSNPs) in human NF2 gene responsible for neurofibromatosis type 2 (NF2). J. Biomol. Struct. Dyn. 2020, 39, 7044–7055. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Das, P.; Purohit, R. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput. Biol. Med. 2021, 128, 104117. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Kumar Bhardwaj, V.; Singh, R.; Rajendran, V.; Purohit, R.; Kumar, S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021, 346. [Google Scholar] [CrossRef]
- Ensembl Ensembl. Available online: https://useast.ensembl.org/index.html (accessed on 10 October 2021).
- UniProt UniProtKB—Q9H3D4 (P63_HUMAN). Available online: https://www.uniprot.org/uniprot/Q9H3D4 (accessed on 10 October 2021).
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Calabrese, R.; Casadio, R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics 2005, 21, ii54–ii58. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant 2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Lapcevic, R.; Romero, A.E.; Yoon, M.; Das, J.; Beltrán, J.F.; Mort, M.; Stenson, P.D.; Cooper, D.N.; Paccanaro, A.; et al. mutation3D: Cancer Gene Prediction through Atomic Clustering of Coding Variants in the Structural Proteome. Hum. Mutat. 2016, 37, 447–456. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Hossain, M.S.; Roy, A.S.; Islam, M.S. In silico analysis predicting effects of deleterious SNPs of human RASSF5 gene on its structure and functions. Sci. Rep. 2020, 10, 14542. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pK a prediction with hydrogen bonding network optimization. In Computational Drug Discovery and Design; Springer: Berlin, Germany, 2012; pp. 405–421. [Google Scholar]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.A.; Bui, N.H.B.; Rajapakshe, K.; Wong, J.; Gunaratne, P.; Tsai, K.Y.; Coarfa, C.; Flores, E.R. Distinct TP63 isoform-driven transcriptional signatures predict tumor progression and clinical outcomes. Cancer Res. 2018, 78, 451–462. [Google Scholar] [CrossRef]
- Serber, Z.; Lai, H.C.; Yang, A.; Ou, H.D.; Sigal, M.S.; Kelly, A.E.; Darimont, B.D.; Duijf, P.H.G.; van Bokhoven, H.; McKeon, F.; et al. A C-Terminal Inhibitory Domain Controls the Activity of p63 by an Intramolecular Mechanism. Mol. Cell. Biol. 2002, 22, 8601–8611. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Chen, C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ. 2008, 15, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Ohba, M.; Kawahara, C.; Ishioka, C.; Kanamaru, R.; Katoh, I.; Ikawa, Y.; Nimura, Y.; Nakagawara, A.; Obinata, M. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 1998, 4, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Ianakiev, P.; Kilpatrick, M.W.; Toudjarska, I.; Basel, D.; Beighton, P.; Tsipouras, P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am. J. Hum. Genet. 2000, 67, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, G.; Hadj-Rabia, S.; Faivre, L.; Sarafan-Vasseur, N.; Frébourg, T. The Rapp-Hodgkin syndrome results from mutations of the TP63 gene. Eur. J. Hum. Genet. 2003, 11, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gonzalez, A.; Rodriguez-Fontenla, C.; Carracedo, A. De novo mutations (DNMs) in autism spectrum disorder (ASD): Pathway and network analysis. Front. Genet. 2018, 9, 406. [Google Scholar] [CrossRef]
- Rodriguez-Casado, A. In silico investigation of functional nsSNPs—An approach to rational drug design. Res. Rep. Med. Chem. 2012, 2, 31–42. [Google Scholar] [CrossRef]
- Hicks, S.; Wheeler, D.A.; Plon, S.E.; Kimmel, M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011, 32, 661–668. [Google Scholar] [CrossRef]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Peleg, O.; Choi, J.-M.; Shakhnovich, E.I. Evolution of specificity in protein-protein interactions. Biophys. J. 2014, 107, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, H.; Nussinov, R.; Ma, B. Protein charge and mass contribute to the spatio-temporal dynamics of protein–protein interactions in a minimal proteome. Proteomics 2013, 13, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.K.; Purohit, R. A new insight into protein-protein interactions and the effect of conformational alterations in PCNA. Int. J. Biol. Macromol. 2020, 148, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mohammad, T.; Samarth, N.; Hussain, A.; Rehman, M.T.; Islam, A.; Alajmi, M.F.; Singh, S.; Hassan, M.I. Structural genomics approach to investigate deleterious impact of nsSNPs in conserved telomere maintenance component 1. Sci. Rep. 2021, 11, 10202. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Ali, M.C.; Rana, M.L.; Munni, Y.A.; Barua, L.; Jahan, I.; Haque, M.F.; Hannan, M.A.; Moon, I.S. Computational SNP analysis and molecular simulation revealed the most deleterious missense variants in the NBD1 domain of human ABCA1 transporter. Int. J. Mol. Sci. 2020, 21, 7606. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Conformational changes of the p53-binding cleft of MDM2 revealed by molecular dynamics simulations. Biopolym. Orig. Res. Biomol. 2006, 83, 365–373. [Google Scholar] [CrossRef]
- Owji, H.; Eslami, M.; Nezafat, N.; Ghasemi, Y. In Silico Elucidation of Deleterious Non-synonymous SNPs in SHANK3, the Autism Spectrum Disorder Gene. J. Mol. Neurosci. 2020, 70, 1649–1667. [Google Scholar] [CrossRef]
- Van Bokhoven, H.; Hamel, B.C.J.; Bamshad, M.; Sangiorgi, E.; Gurrieri, F.; Duijf, P.H.G.; Vanmolkot, K.R.J.; Van Beusekom, E.; Van Beersum, S.E.C.; Celli, J.; et al. p63 gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am. J. Hum. Genet. 2001, 69, 481–492. [Google Scholar] [CrossRef]
- Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017, 7, 170019. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Muñoz Aguirre, M. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar]
- Westra, H.-J.; Franke, L. From genome to function by studying eQTLs. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1896–1902. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | rsID (dbSNP) | Domain Function | Methods | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutpred2 | PROVEAN | ClinVar | PhD-SNP | PANTHER | SNPs&GO | ||||||||
| Mdscore | Mutpred2 Impact | Score | Impact | Result | Prediction | Probability | Prediction | Probability | Prediction | Probability | |||
| R408C | rs1282887680 | Oligomerization | 0.662 | −7.064 | Deleterious | not found | Disease | 0.771 | Disease | 0.927 | Disease | 0.996 | |
| R408H | rs751698974 | Oligomerization | 0.508 | −4.461 | Deleterious | not found | Disease | 0.767 | Disease | 0.83 | Disease | 0.996 | |
| R376C | rs757536818 | Interaction with HIPK21 | 0.355 | −3.65 | Deleterious | not found | Disease | 0.716 | Neutral | 0.477 | Disease | 0.996 | |
| C347F | rs1064793282 | DNA binding domain | 0.932 | Gain of Strand (Pr = 0.27 | P = 0.03) | −10.073 | Deleterious | Pathogenic | Disease | 0.91 | Disease | 0.975 | Disease | 0.997 |
| D351G | rs121908844 | DNA binding domain | 0.863 | −6.41 | Deleterious | Pathogenic | Disease | 0.836 | Disease | 0.916 | Disease | 0.997 | |
| D355N | rs1553857889 | DNA binding domain/Interaction with HIPK21 | 0.706 | −3.512 | Deleterious | Pathogenic | Neutral | 0.337 | Disease | 0.675 | Disease | 0.988 | |
| G349E | rs866267914 | DNA binding domain | 0.852 | Gain-Intrinsic disorder P = 0.04 Loss of Strand P = 0.02 | −7.342 | Deleterious | Pathogenic | Disease | 0.661 | Disease | 0.955 | Disease | 0.987 |
| R266Q | rs121908849 | DNA binding domain | 0.807 | Loss of Strand P = 0.02 Altered Stability P = 0.01 | −3.612 | Deleterious | Pathogenic | Disease | 0.806 | Disease | 0.924 | Disease | 0.994 |
| R318H | rs121908840 | DNA binding domain | 0.725 | Loss-ADP-ribosylation at R318 P = 0.03 | −4.645 | Deleterious | Pathogenic | Disease | 0.885 | Disease | 0.959 | Disease | 0.995 |
| R319H | rs886039442 | DNA binding domain | 0.742 | −4.627 | Deleterious | Pathogenic | Disease | 0.865 | Disease | 0.948 | Disease | 0.995 | |
| R337Q | rs113993967 | DNA binding domain | 0.861 | Gain-Strand P = 0.02 Gain-ADP-ribosylation at R338 P = 0.05 Gain-Pyrrolidone carboxylic acid at R337 P = 0.02 | −3.618 | Deleterious | Pathogenic | Disease | 0.8 | Disease | 0.901 | Disease | 0.995 |
| R343Q | rs121908841 | DNA binding domain | 0.801 | Gain of Strand P = 0.03; Altered Stability P = 0.02 | −3.663 | Deleterious | Pathogenic | Disease | 0.855 | Disease | 0.902 | Disease | 0.996 |
| R379C | rs761885185 | Interaction with HIPK21 | 0.515 | Loss- Intrinsic disorder P = 0.02); Loss-Phosphorylation at T382 P = 0.01; Loss-Acetylation at K375 | P = 0.01; Altered Disordered interface P = 0.04 | −2.648 | Deleterious | Uncertain significance | Neutral | 0.424 | Disease | 0.602 | Disease | 0.995 |
| R379H | rs765502786 | Interaction with HIPK21 | 0.312 | −1.476 | Neutral | Uncertain significance | Neutral | 0.227 | Neutral | 0.413 | Disease | 0.989 | |
| L562R | rs774221257 | SAM | 0.896 | Altered Transmembrane protein P = 9.7 × 10−5 Altered Ordered interface P = 0.02. Altered Stability P = 0.03. Loss-Sulfation at Y564 P = 0.03 | −2.328 | Neutral | not found | Disease | 0.787 | Disease | 0.517 | Disease | 0.998 |
| R647H | rs774550896 | Transactivation inhibition | 0.834 | Altered Metal binding P = 2.9 × 10−3. Altered DNA binding P = 1.2 × 10−3; Altered Disordered interface P = 0.04). Loss-Proteolytic cleavage at R643 P = 0.02. Altered Transmembrane protein P = 0.03. Altered Stability P = 0.04 | −2.062 | Neutral | not found | Disease | 0.761 | Disease | 0.743 | Disease | 0.997 |
| R655Q | rs764601563 | Transactivation inhibition | 0.656 | Altered Disordered interface P = 0.04. Altered Metal binding P = 0.03. Altered DNA binding P = 0.03. Altered Transmembrane protein P = 0.05 | −1.246 | Neutral | not found | Disease | 0.755 | Disease | 0.591 | Disease | 0.996 |
| SNP | Stability | |
|---|---|---|
| R408C | −1.01 | Decreased |
| R408H | −1.38 | |
| C347F | −0.48 | |
| D351G | −1.64 | |
| D355N | −1.49 | |
| G349E | −1.48 | |
| R266Q | −1.03 | |
| R318H | −1.3 | |
| R319H | −1.38 | |
| R337Q | −0.91 | |
| R343Q | −0.99 | |
| R379C | −0.41 | |
| R379C | −0.14 | |
| L562R | −1.84 | |
| R647H | −1.97 | |
| R655Q | −1.96 |
| SNPs | Size & Charge | Characteristics & Features | |
|---|---|---|---|
| Wild-Type | Mutant | ||
| R376C | Large & (+ve) | Small & (0) | Hydrophobicity: High. Effect: High. Protean folding: Affected. Loss of interaction: High & distributed; |
| R4(0)8C | Large & (+ve) | Small & (0) | |
| R4(0)8H | Large & (+ve) | Small & (0) | |
| C347F | Small & (0) | Large & (0) | |
| D351G | Large & (−ve) | Small & (0) | |
| D355N | Large & (−ve) | Small & (0) | |
| G349E | Small & (0) | Large & (−ve) | |
| R266Q | Large & (+ve) | Small & (0) | |
| R318H | Large & (+ve) | Small & (0) | |
| R319H | Large & (+ve) | Small & (0) | |
| R337Q | Large & (+ve) | Small & (0) | |
| R343Q | Large & (+ve) | Small & (0) | |
| R379C | Large & (+ve) | Small & (0) | |
| R379C | Large & (+ve) | Small & (0) | |
| L562R | Small & +(0) | Large & (+ve) | |
| R647H | Large & (+ve) | Small & (+ve) | |
| R655Q | Large & (+ve) | Small & (+ve) | |
| Wild-Type | Mutant | |||
|---|---|---|---|---|
| Residue | Binding Affinity | Residue | Binding Affinity | Binding Affinity Change |
| R266 | −6.2 | Q266 | −5.9 | Decrease |
| R318 | −5.3 | H318 | −6.2 | Increase |
| R319 | −5.8 | H319 | −5.4 | Decrease |
| R337 | −5.5 | Q337 | −5.8 | Increase |
| R343 | −5.8 | Q343 | −5.8 | Neutral |
| C347 | −6 | F347 | −5.6 | Decrease |
| G349 | −6.4 | E349 | −5.8 | Decrease |
| D351 | −5.6 | G351 | −5.4 | Decrease |
| D355 | 5 | N355 | −5.8 | Increase |
| Gene Symbol | Variant ID | SNP ID (Non-Coding) | p-Value | NES | Single Tissue eQTL |
|---|---|---|---|---|---|
| TP63 | chr3_189638472_T_C_b38 | rs4488809 | 6.5 × 10−7 | 0.23 | Lung |
| TP63 | chr3_189638472_T_C_b38 | rs4488809 | 1.7 × 10−5 | 0.13 | Nerve-Tibial |
| TP63 | chr3_189664468_A_G_b38 | rs6774934 | 5.5 × 10−5 | −0.34 | Heart-Left Ventricle |
| TP63 | chr3_189664468_A_G_b38 | rs6774934 | 6.5 × 10−5 | −0.20 | Nerve-Tibial |
| TP63 | chr3_189672911_G_C_b38 | rs6794898 | 1.3 × 10−5 | 0.20 | Lung |
| TP63 | chr3_189710792_T_G_b38 | rs79155799 | 6.5 × 10−7 | −0.17 | Nerve-Tibial |
| TP63 | chr3_189721190_A_G_b38 | rs4687090 | 1.7 × 10−5 | −0.19 | Nerve-Tibial |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akter, S.; Hossain, S.; Ali, M.A.; Hosen, M.I.; Shekhar, H.U. Comprehensive Characterization of the Coding and Non-Coding Single Nucleotide Polymorphisms in the Tumor Protein p63 (TP63) Gene Using In Silico Tools. Biomolecules 2021, 11, 1733. https://doi.org/10.3390/biom11111733
Akter S, Hossain S, Ali MA, Hosen MI, Shekhar HU. Comprehensive Characterization of the Coding and Non-Coding Single Nucleotide Polymorphisms in the Tumor Protein p63 (TP63) Gene Using In Silico Tools. Biomolecules. 2021; 11(11):1733. https://doi.org/10.3390/biom11111733
Chicago/Turabian StyleAkter, Shamima, Shafaat Hossain, Md. Ackas Ali, Md. Ismail Hosen, and Hossain Uddin Shekhar. 2021. "Comprehensive Characterization of the Coding and Non-Coding Single Nucleotide Polymorphisms in the Tumor Protein p63 (TP63) Gene Using In Silico Tools" Biomolecules 11, no. 11: 1733. https://doi.org/10.3390/biom11111733