
Rare Does Not Mean Worthless: How Rare Diseases Have Shaped Neurodevelopment Research in the NGS Era


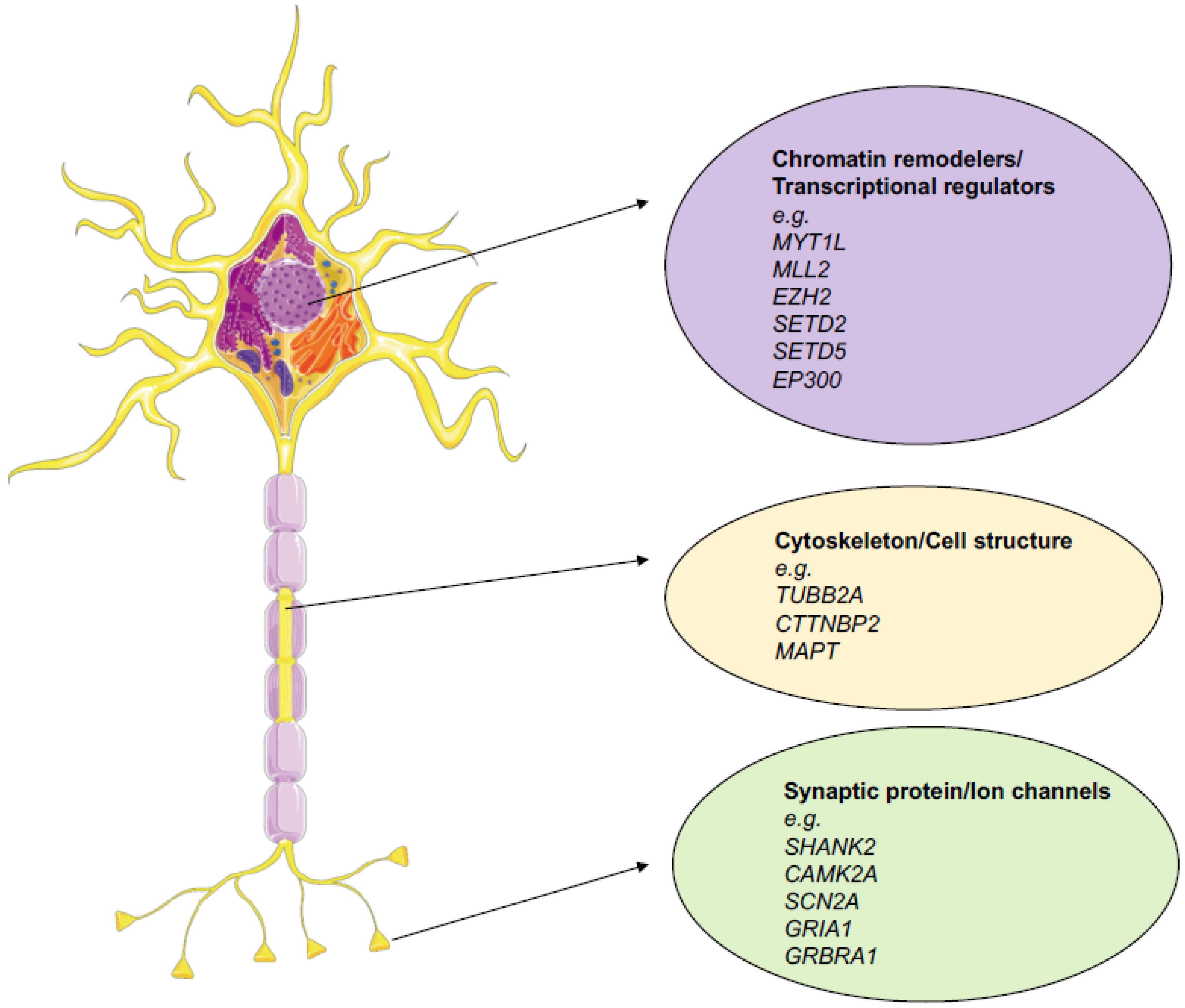{kind=link}
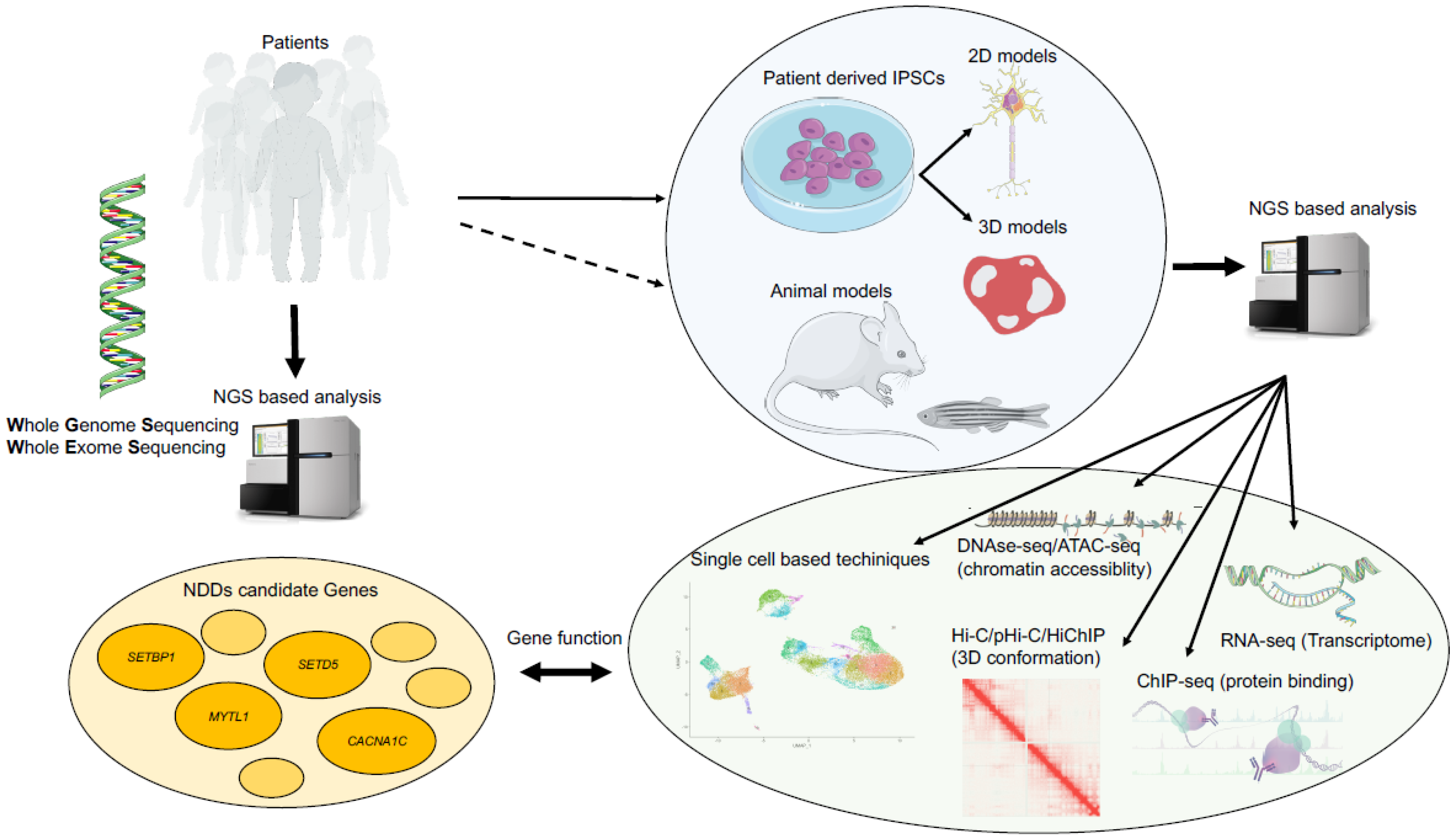{kind=link}
Abstract
:1. Introduction
2. NGS Impact on NDD Diagnosis
2.1. Whole Exome Sequencing and the Discoveries of New Variants
2.2. Sequencing Contribution in Guiding Biological Questions
2.3. NGS and Non-Coding Variant Interpretation
3. NGS in Experimental Modelling of NDDs
3.1. Transcriptomic Analysis
3.2. Epigenome Studies
3.3. D Chromatin Structure Investigation
3.4. Single-Cell Approaches
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Hampshire, D.J.; Pattison, L.; Springell, K.; Jafri, H.; Corry, P.; Mannon, J.; Rashid, Y.; Crow, Y.; Bond, J.; et al. Autosomal recessive primary microcephaly: An analysis of locus heterogeneity and phenotypic variation. J. Med. Genet. 2002, 39, 718–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Nigro, C.; Chong, S.S.; Smith, A.C.M.; Dobyns, W.B.; Carrozzo, R.; Ledbetter, D.H. Point Mutations and an Intragenic Deletion in LIS1, the Lissencephaly Causative Gene in Isolated Lissencephaly Sequence and Miller-Dieker Syndrome. Hum. Mol. Genet. 1997, 6, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Des Portes, V.; Pinard, J.M.; Billuart, P.; Vinet, M.C.; Koulakoff, A.; Carrié, A.; Gelot, A.; Dupuis, E.; Motte, J.; Berwald-Netter, Y.; et al. A Novel CNS Gene Required for Neuronal Migration and Involved in X-Linked Subcortical Laminar Heterotopia and Lissencephaly Syndrome. Cell 1998, 92, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [Green Version]
- Gilissen, C.; Arts, H.H.; Hoischen, A.; Spruijt, L.; Mans, D.A.; Arts, P.; Van Lier, B.; Steehouwer, M.; Van Reeuwijk, J.; Kant, S.G.; et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 2010, 87, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Hoischen, A.; Van Bon, B.W.M.; Gilissen, C.; Arts, P.; Van Lier, B.; Steehouwer, M.; De Vries, P.; De Reuver, R.; Wieskamp, N.; Mortier, G.; et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010, 42, 483–485. [Google Scholar] [CrossRef]
- Schinzel, A.; Giedion, A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformations in sibs. Am. J. Med. Genet. 1978, 1, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Sensenbrenner, J.A.; Dorst, J.P.; Owens, R.P. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig. Artic. Ser. 1975, 11, 372–379. [Google Scholar] [PubMed]
- Miller, M.; Fineman, R.; Smith, D.W. Postaxial acrofacial dysostosis syndrome. J. Pediatr. 1979, 95, 970–975. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; MacKenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; Di Lullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Iossifov, I.; O/’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Deliu, E.; Arecco, N.; Morandell, J.; Dotter, C.P.; Contreras, X.; Girardot, C.; Käsper, E.L.; Kozlova, A.; Kishi, K.; Chiaradia, I.; et al. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 2018, 21, 1717–1727. [Google Scholar] [CrossRef]
- Sessa, A.; Fagnocchi, L.; Mastrototaro, G.; Massimino, L.; Zaghi, M.; Indrigo, M.; Cattaneo, S.; Martini, D.; Gabellini, C.; Pucci, C.; et al. SETD5 Regulates Chromatin Methylation State and Preserves Global Transcriptional Fidelity during Brain Development and Neuronal Wiring. Neuron 2019, 104, 271–289. [Google Scholar] [CrossRef]
- Moore, S.M.; Seidman, J.S.; Ellegood, J.; Gao, R.; Savchenko, A.; Troutman, T.D.; Abe, Y.; Stender, J.; Lee, D.; Wang, S.; et al. Setd5 haploinsufficiency alters neuronal network connectivity and leads to autistic-like behaviors in mice. Transl. Psychiatry 2019, 9, 24. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.-B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; He, Y.; Rehman, A.U.; Kong, Y.; Hong, S.; Ding, G.; Yalamanchili, H.K.; Wan, Y.-W.; Paul, B.; Wang, C.; et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus–CA3 projection. Nat. Neurosci. 2019, 22, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Kareta, M.S.; Chanda, S.; Ahlenius, H.; Perotti, N.; Zhou, B.; Grieder, S.D.; Ge, X.; Drake, S.; Euong Ang, C.; et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 2017, 544, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.N.; Eichler, E.E. The Role of De Novo Noncoding Regulatory Mutations in Neurodevelopmental Disorders. Trends Neurosci. 2019, 42, 115–127. [Google Scholar] [CrossRef] [PubMed]
- de la Torre-Ubieta, L.; Stein, J.L.; Won, H.; Opland, C.K.; Liang, D.; Lu, D.; Geschwind, D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172, 289–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevino, A.E.; Sinnott-Armstrong, N.; Andersen, J.; Yoon, S.-J.; Huber, N.; Pritchard, J.K.; Chang, H.Y.; Greenleaf, W.J.; Pașca, S.P. Chromatin accessibility dynamics in a model of human forebrain development. Science 2020, 367, eaay1645. [Google Scholar] [CrossRef]
- Trevino, A.E.; Müller, F.; Andersen, J.; Sundaram, L.; Kathiria, A.; Shcherbina, A.; Farh, K.; Chang, H.Y.; Pașca, A.M.; Kundaje, A.; et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 2021, 184, 5053–5069. [Google Scholar] [CrossRef]
- Ziffra, R.S.; Kim, C.N.; Ross, J.M.; Wilfert, A.; Turner, T.N.; Haeussler, M.; Casella, A.M.; Przytycki, P.F.; Keough, K.C.; Shin, D.; et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 2021, 598, 205–213. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Furey, T.S. Identifying and Characterizing Regulatory Sequences in the Human Genome with Chromatin Accessibility Assays. Genes 2012, 3, 651–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, N. The ENCODE project. Nat. Methods 2012, 9, 1046. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.N.; Hormozdiari, F.; Duyzend, M.H.; McClymont, S.A.; Hook, P.W.; Iossifov, I.; Raja, A.; Baker, C.; Hoekzema, K.; Stessman, H.A.; et al. Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am. J. Hum. Genet. 2016, 98, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.N.; Coe, B.P.; Dickel, D.E.; Hoekzema, K.; Nelson, B.J.; Zody, M.C.; Kronenberg, Z.N.; Hormozdiari, F.; Raja, A.; Pennacchio, L.A.; et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 2017, 171, 710–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; FitzPatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Kleiber, M.L.; Whitney, J.; Maile, M.S.; Hong, O.; Chapman, T.R.; Tan, S.; Tandon, P.; et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 2018, 360, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Yuen, R.K.C.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. npj Genom. Med. 2016, 1, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Steffenburg, S.; Gillberg, C.; Hellgren, L.; Andersson, L.; Gillberg, I.C.; Jakobsson, G.; Bohman, M. A Twin Study of Autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry 1989, 30, 405–416. [Google Scholar] [CrossRef]
- Stassen, M.J.; Bailey, D.; Nelson, S.; Chinwalla, V.; Harte, P.J. The Drosophilatrithorax proteins contain a novel variant of the nuclear receptor type DNA binding domain and an ancient conserved motif found in other chromosomal proteins. Mech. Dev. 1995, 52, 209–223. [Google Scholar] [CrossRef]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.-X.; et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratten, J.; Wray, N.R.; Peyrot, W.J.; McGrath, J.J.; Visscher, P.M.; Goddard, M.E. Risk of psychiatric illness from advanced paternal age is not predominantly from de novo mutations. Nat. Genet. 2016, 48, 718–724. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39, S37–S42. [Google Scholar] [CrossRef] [PubMed]
- de Kok, Y.J.M.; Merkx, G.F.M.; van der Maarel, S.M.; Huber, I.; Malcolm, S.; Ropers, H.-H.; Cremers, F.P.M. A duplication/paracentric inversion associated with familial X-linked deafness (DFN3) suggests the presence of a regulatory element more than 400 kb upstream of the POU3F4 gene. Hum. Mol. Genet. 1995, 4, 2145–2150. [Google Scholar] [CrossRef] [Green Version]
- Lettice, L.A.; Heaney, S.J.H.; Purdie, L.A.; Li, L.; de Beer, P.; Oostra, B.A.; Goode, D.; Elgar, G.; Hill, R.E.; de Graaff, E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003, 12, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.J.; Huang, G.-J.; Cheung, A.F.P.; Era, T.; Nishikawa, S.-I.; Bikoff, E.K.; Molnár, Z.; Robertson, E.J.; Groszer, M. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008, 22, 2479–2484. [Google Scholar] [CrossRef] [Green Version]
- Sessa, A.; Mao, C.; Hadjantonakis, A.-K.; Klein, W.H.; Broccoli, V. Tbr2 Directs Conversion of Radial Glia into Basal Precursors and Guides Neuronal Amplification by Indirect Neurogenesis in the Developing Neocortex. Neuron 2008, 60, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baala, L.; Briault, S.; Etchevers, H.C.; Laumonnier, F.; Natiq, A.; Amiel, J.; Boddaert, N.; Picard, C.; Sbiti, A.; Asermouh, A.; et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007, 39, 454–456. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Avsec, Ž.; Weilert, M.; Shrikumar, A.; Krueger, S.; Alexandari, A.; Dalal, K.; Fropf, R.; McAnany, C.; Gagneur, J.; Kundaje, A.; et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 2021, 53, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodin, R.E.; Dou, Y.; Kwon, M.; Sherman, M.A.; D’Gama, A.M.; Doan, R.N.; Rento, L.M.; Girskis, K.M.; Bohrson, C.L.; Kim, S.N.; et al. The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci. 2021, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Lachman, H.M.; Zheng, D. Transcriptomics Analysis of iPSC-derived Neurons and Modeling of Neuropsychiatric Disorders HHS Public Access. Mol. Cell Neurosci. 2016, 73, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ziller, M.; Spengler, D. Progress in iPSC-Based Modeling of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 4896. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Pedrosa, E.; Shah, A.; Hrabovsky, A.; Maqbool, S.; Zheng, D.; Lachman, H.M. RNA-Seq of Human Neurons Derived from iPS Cells Reveals Candidate Long Non-Coding RNAs Involved in Neurogenesis and Neuropsychiatric Disorders. PLoS ONE 2011, 6, e23356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Q.; Habegger, L.; Noisa, P.; Szekely, A.; Qiu, C.; Hutchison, S.; Raha, D.; Egholm, M.; Lin, H.; Weissman, S.; et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 5254–5259. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, W.; Jiang, T. Inference of Isoforms from Short Sequence Reads. J. Comput. Biol. 2011, 18, 305. [Google Scholar] [CrossRef] [PubMed]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 2015, 20, 1350–1365. [Google Scholar] [CrossRef] [Green Version]
- Marchetto, M.; Belinson, H.; Tian, Y.; Freitas, B.; Fu, C.; Vadodaria, K.; Beltrao-Braga, P.; Trujillo, C.; Mendes, A.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2016, 22, 820–835. [Google Scholar] [CrossRef]
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Commun. 2013, 14, 347–359. [Google Scholar] [CrossRef]
- Banfi, F.; Rubio, A.; Zaghi, M.; Massimino, L.; Fagnocchi, G.; Bellini, E.; Luoni, M.; Cancellieri, C.; Bagliani, A.; Di Resta, C.; et al. SETBP1 accumulation induces P53 inhibition and genotoxic stress in neural progenitors underlying neurodegeneration in Schinzel-Giedion syndrome. Nat. Commun. 2021, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Chailangkarn, T.; Trujillo, C.A.; Freitas, B.C.; Hrvoj-Mihic, B.; Herai, R.H.; Yu, D.X.; Brown, T.T.; Marchetto, M.C.; Bardy, C.; McHenry, L.; et al. A human neurodevelopmental model for Williams syndrome. Nature 2016, 536, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, J.L.; de la Torre-Ubieta, L.; Tian, Y.; Parikshak, N.N.; Hernández, I.A.; Marchetto, M.C.; Baker, D.K.; Lu, D.; Hinman, C.R.; Lowe, J.K.; et al. A Quantitative Framework to Evaluate Modeling of Cortical Development by Neural Stem Cells. Neuron 2014, 83, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.S.; Gigek, C.O.; Rosenfeld, J.A.; Diallo, A.B.; Maussion, G.; Chen, G.G.; Vaillancourt, K.; Lopez, J.P.; Crapper, L.; Poujol, R.; et al. Molecular Convergence of Neurodevelopmental Disorders. Am. J. Hum. Genet. 2014, 95, 490–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Radford, J.C.; Ragusa, M.J.; Shea, K.L.; McKercher, S.R.; Zaremba, J.D.; Soussou, W.; Nie, Z.; Kang, Y.-J.; Nakanishi, N.; et al. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 9397–9402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.-A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Melo, U.S.; Schöpflin, R.; Acuna-Hidalgo, R.; Mensah, M.A.; Fischer-Zirnsak, B.; Holtgrewe, M.; Klever, M.K.; Türkmen, S.; Heinrich, V.; Pluym, I.D.; et al. Hi-C Identifies Complex Genomic Rearrangements and TAD-Shuffling in Developmental Diseases. Am. J. Hum. Genet. 2020, 106, 872–884. [Google Scholar] [CrossRef]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.Y.; Hwang, J.H.; Lee, J.Y.; Cho, A.N.; Lee, A.J.; Jung, I.; Cho, S.W.; Kim, L.K.; Kim, Y.J. Chromatin interaction changes during the ipsc-npc model to facilitate the study of biologically significant genes involved in differentiation. Genes 2020, 11, 1176. [Google Scholar] [CrossRef]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, 4311. [Google Scholar] [CrossRef] [Green Version]
- Mumbach, M.R.; Satpathy, A.T.; Boyle, E.A.; Dai, C.; Gowen, B.G.; Cho, S.W.; Nguyen, M.L.; Rubin, A.J.; Granja, J.M.; Kazane, K.R.; et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017, 49, 1602–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019, 51, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Platt, R.; Treutlein, B. Mapping human cell phenotypes to genotypes with single-cell genomics. Science 2019, 365, 1401–1405. [Google Scholar] [CrossRef]
- Cuomo, A.S.E.; Seaton, D.D.; McCarthy, D.J.; Martinez, I.; Bonder, M.J.; Garcia-Bernardo, J.; Amatya, S.; Madrigal, P.; Isaacson, A.; Buettner, F.; et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Ha, T.W.; Lee, M.R. Single-Cell Transcriptome Analysis as a Promising Tool to Study Pluripotent Stem Cell Reprogramming. Int. J. Mol. Sci. 2021, 22, 5988. [Google Scholar] [CrossRef]
- Nehme, R.; Zuccaro, E.; Ghosh, S.D.; Li, C.; Sherwood, J.L.; Pietilainen, O.; Barrett, L.E.; Limone, F.; Worringer, K.A.; Kommineni, S.; et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 2018, 23, 2509–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.-K.; Micali, N.; Wang, Y.; Colantuoni, C.; Straub, R.E.; Hoeppner, D.J.; et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brancati, G.; Treutlein, B.; Camp, J.G. Resolving Neurodevelopmental and Vision Disorders Using Organoid Single-Cell Multi-omics. Neuron 2020, 107, 1000–1013. [Google Scholar] [CrossRef]
- Bock, C.; Boutros, M.; Camp, J.G.; Clarke, L.; Clevers, H.; Knoblich, J.A.; Liberali, P.; Regev, A.; Rios, A.C.; Stegle, O.; et al. The Organoid Cell Atlas. Nat. Biotechnol. 2020, 39, 13–17. [Google Scholar] [CrossRef]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesisin a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Yang, S.M.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G.L. Generation of human brain region–specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 2018, 13, 565–580. [Google Scholar] [CrossRef]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, A.C.; Kyrousi, C.; Einsiedler, M.; Burtscher, I.; Drukker, M.; Markie, D.M.; Kirk, E.P.; Götz, M.; Robertson, S.P.; Cappello, S. Mob2 insufficiency disrupts neuronal migration in the developing cortex. Front. Cell. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.C.; Kyrousi, C.; Klaus, J.; Leventer, R.J.; Kirk, E.P.; Fry, A.; Pilz, D.T.; Morgan, T.; Jenkins, Z.A.; Drukker, M.; et al. A Primate-Specific Isoform of PLEKHG6 Regulates Neurogenesis and Neuronal Migration. Cell Rep. 2018, 25, 2729–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, J.; Kanton, S.; Kyrousi, C.; Ayo-Martin, A.C.; Di Giaimo, R.; Riesenberg, S.; O’Neill, A.C.; Camp, J.G.; Tocco, C.; Santel, M.; et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 2019, 25, 561–568. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-dependent dysregulation of GABA/glutamate neurondifferentiation in autism spectrum disorders. Cell 2015, 162, 375. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Kang, E.; Yu, C.; Qian, X.; Jacob, F.; Yu, C.; Mao, M.; Poon, R.Y.C.; Kim, J.; Song, H.; et al. DISC1 Regulates Neurogenesis via Modulating Kinetochore Attachment of Ndel1/Nde1 during Mitosis. Neuron 2017, 96, 1204. [Google Scholar] [CrossRef] [Green Version]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. Human cerebral organoids reveal deficits in neurogenesis and neuronal migration in MeCP2-deficient neural progenitors. Mol. Psychiatry 2018, 23, 791. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Zhou, Y.; Li, Y.; Han, Y.; Xu, J.; Niu, W.; Li, Z.; Liu, S.; Feng, H.; Huang, W.; et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 2021, 24, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Bowles, K.R.; Silva, M.C.; Whitney, K.; Bertucci, T.; Berlind, J.E.; Lai, J.D.; Garza, J.C.; Boles, N.C.; Mahali, S.; Strang, K.H.; et al. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 2021, 184, 4547–4563. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchís-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef]
- Callaway, E.M.; Dong, H.-W.; Ecker, J.R.; Hawrylycz, M.J.; Huang, Z.J.; Lein, E.S.; Ngai, J.; Osten, P.; Ren, B.; Tolias, A.S.; et al. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 2021, 598, 86–102. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [Green Version]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaghi, M.; Banfi, F.; Bellini, E.; Sessa, A. Rare Does Not Mean Worthless: How Rare Diseases Have Shaped Neurodevelopment Research in the NGS Era. Biomolecules 2021, 11, 1713. https://doi.org/10.3390/biom11111713
Zaghi M, Banfi F, Bellini E, Sessa A. Rare Does Not Mean Worthless: How Rare Diseases Have Shaped Neurodevelopment Research in the NGS Era. Biomolecules. 2021; 11(11):1713. https://doi.org/10.3390/biom11111713
Chicago/Turabian StyleZaghi, Mattia, Federica Banfi, Edoardo Bellini, and Alessandro Sessa. 2021. "Rare Does Not Mean Worthless: How Rare Diseases Have Shaped Neurodevelopment Research in the NGS Era" Biomolecules 11, no. 11: 1713. https://doi.org/10.3390/biom11111713