
Data-Driven Analysis of Fluorination of Ligands of Aminergic G Protein Coupled Receptors

 , , and
, , and 
Abstract
:1. Introduction
2. Materials, Methods, and Analysis Concepts
2.1. Compounds and Activity Data
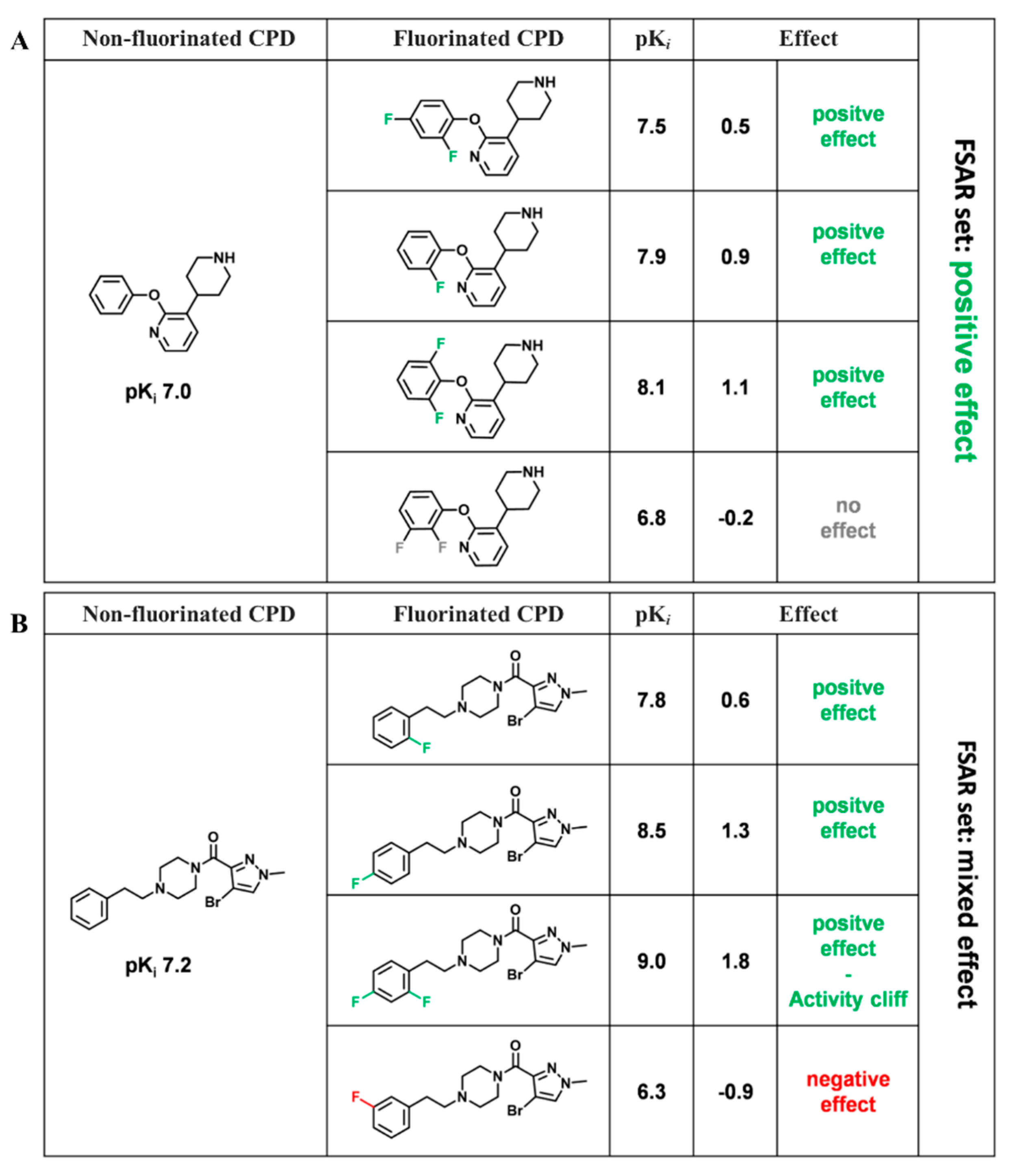
2.2. Fluorine-Dependent Analogue Sets
- (i)
- No effect: MIN ΔpPot > −0.3 and MAX ΔpPot < 0.3
- (ii)
- Positive effect: MIN ΔpPot ≥ −0.3 and MAX ΔpPot > 0.3
- (iii)
- Negative effect: MAX ΔpPot ≤ −0.3 and MIN ΔpPot < 0.3
- (iv)
- Mixed effect: MIN ΔpPot < −0.3 and MAX ΔpPot > 0.3
- (v)
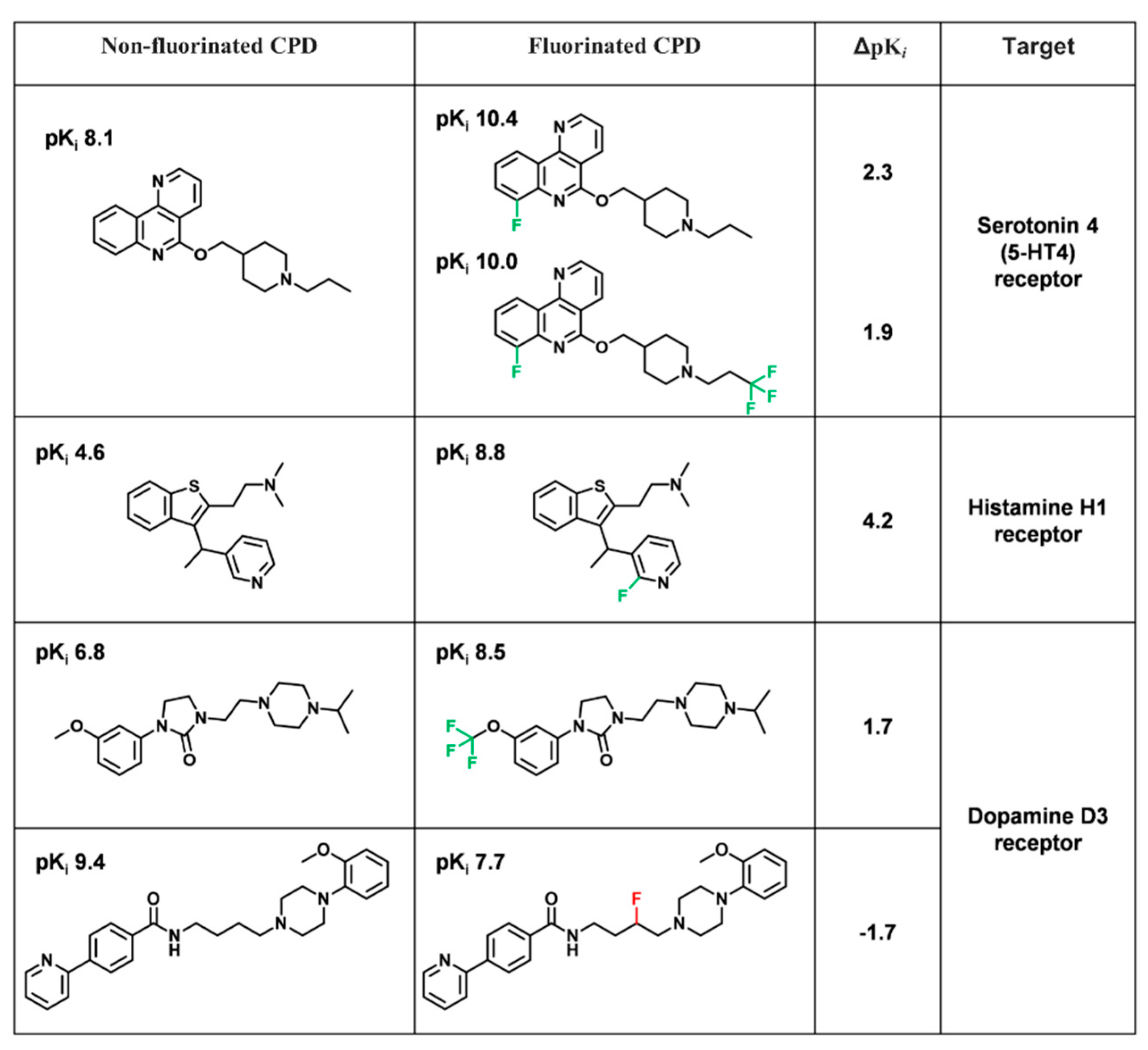
- Inconclusive effect: MIN ΔpPot ≤ −1.7 and MAX ΔpPot ≥ 1.7
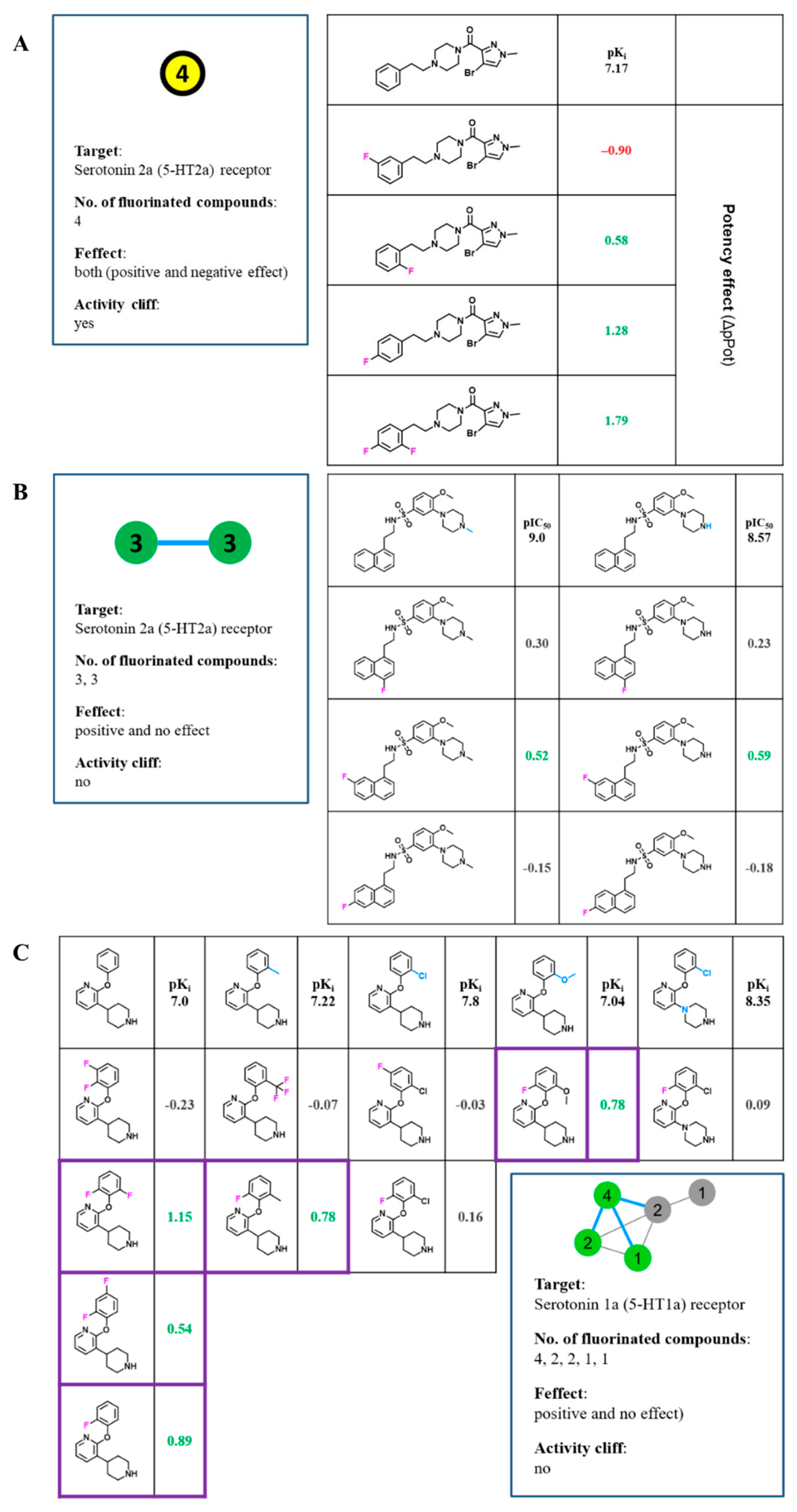
2.3. Activity Cliffs
2.4. Matched Molecular Pairs
2.5. MMP Networks
- (i).
- No effect: No effects for all targets (grey).
- (ii).
- Positive effect: Only positive effects (possibly in combination with no effects) (green).
- (iii).
- Negative effect: Only negative effects (possibly in combination with no effects) (red).
- (iv).
- Mixed effect: Combinations of negative, positive, and mixed effects (possibly in combination with no effects) (yellow).
3. Results and Discussion
3.1. FSAR Sets
3.2. Activity Cliffs in FSAR Sets and Their Interpretation
3.3. MMP Network
3.4. SAR Rules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ursu, O.; Holmes, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Stathias, V.; Nguyen, D.T.; Schürer, S.; Oprea, T. DrugCentral 2018: An update. Nucleic Acids Res. 2019, 47, D963–D970. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Mohammad Nezhady, M.A.; Rivera, J.C.; Chemtob, S. Location Bias as Emerging Paradigm in GPCR Biology and Drug Discovery. iScience 2020, 23, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Bologa, C.G.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Swallow, S. Fluorine in Medicinal Chemistry. In Progress in Medicinal Chemistry; Elsevier B.V.: Edinburgh, UK, 2015; Volume 54, pp. 65–133. ISBN 0306-0012. [Google Scholar]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem.–Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Blakemore, D.C.; Narayanan, A.; Unwalla, R.; Lovering, F.; Denny, R.A.; Zhou, H.; Bunnage, M.E. Fluorine in drug design: A case study with fluoroanisoles. ChemMedChem 2015, 10, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Vass, M.; Podlewska, S.; De Esch, I.J.P.; Bojarski, A.J.; Leurs, R.; Kooistra, A.J.; De Graaf, C. Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J. Med. Chem. 2019, 62, 3784–3839. [Google Scholar] [CrossRef] [PubMed]
- Hussain, J.; Rea, C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 2010, 50, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W.; Sadowski, J. Structure Modification in Chemical Databases. In Chemoinformatics in Drug Discovery; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; Volume 23, pp. 271–285. [Google Scholar]
- Hu, X.; Hu, Y.; Vogt, M.; Stumpfe, D.; Bajorath, J. MMP-cliffs: Systematic identification of activity cliffs on the basis of matched molecular pairs. J. Chem. Inf. Model. 2012, 52, 1138–1145. [Google Scholar] [CrossRef]
- Stumpfe, D.; Hu, Y.; Dimova, D.; Bajorath, J. Recent progress in understanding activity cliffs and their utility in medicinal chemistry. J. Med. Chem. 2014, 57, 18–28. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bajorath, J.; Hu, Y.; Stumpfe, D. Advancing the activity cliff concept. F1000Research 2013, 2, 199. [Google Scholar] [CrossRef] [Green Version]
- Pietruś, W.; Kurczab, R.; Kafel, R.; Machalska, E.; Kalinowska-Tłuścik, J.; Hogendorf, A.; Żylewski, M.; Baranska, M.; Bojarski, A.J. How can fluorine directly and indirectly affect the hydrogen bonding in molecular systems? – A case study for monofluoroanilines. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 252, 119536. [Google Scholar] [CrossRef]
- Dubost, E.; Dumas, N.; Fossey, C.; Magnelli, R.; Butt-Gueulle, S.; Ballandonne, C.; Caignard, D.H.; Dulin, F.; Sopkova De-Oliveira Santos, J.; Millet, P.; et al. Synthesis and structure-affinity relationships of selective high-affinity 5-HT4 receptor antagonists: Application to the design of new potential single photon emission computed tomography tracers. J. Med. Chem. 2012, 55, 9693–9707. [Google Scholar] [CrossRef] [Green Version]
- Moree, W.J.; Jovic, F.; Coon, T.; Yu, J.; Li, B.F.; Tucci, F.C.; Marinkovic, D.; Gross, R.S.; Malany, S.; Bradbury, M.J.; et al. Novel benzothiophene H1-antihistamines for the treatment of insomnia. Bioorganic Med. Chem. Lett. 2010, 20, 2316–2320. [Google Scholar] [CrossRef]
- Micheli, F.; Holmes, I.; Arista, L.; Bonanomi, G.; Braggio, S.; Cardullo, F.; Di Fabio, R.; Donati, D.; Gentile, G.; Hamprecht, D.; et al. Dopamine D3 receptor antagonists: The quest for a potentially selective PET ligand. Part two: Lead optimization. Bioorganic Med. Chem. Lett. 2009, 19, 4011–4013. [Google Scholar] [CrossRef] [PubMed]
- Grundt, P.; Prevatt, K.M.; Cao, J.; Taylor, M.; Floresca, C.Z.; Choi, J.K.; Jenkins, B.G.; Luedtke, R.R.; Newman, A.H. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl) arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: Potential substance abuse therapeutic agents. J. Med. Chem. 2007, 50, 4135–4146. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Ullman, B.; Choi, J.S.K.; Cherrier, M.; Strah-Pleynet, S.; Decaire, M.; Dosa, P.I.; Feichtinger, K.; Teegarden, B.R.; Frazer, J.M.; et al. Synthesis and in vivo evaluation of phenethylpiperazine amides: Selective 5-hydroxytryptamine2A receptor antagonists for the treatment of insomnia. J. Med. Chem. 2010, 53, 5696–5706. [Google Scholar] [CrossRef]
- Blass, B. Sulfonamide Derivatives and Pharmaceutical Applications Thereof. ACS Med. Chem. Lett. 2016, 7, 12–14. [Google Scholar] [CrossRef] [Green Version]
- Dounay, A.B.; Barta, N.S.; Campbell, B.M.; Coleman, C.; Collantes, E.M.; Denny, L.; Dutta, S.; Gray, D.L.; Hou, D.; Iyer, R.; et al. Design, synthesis, and pharmacological evaluation of phenoxy pyridyl derivatives as dual norepinephrine reuptake inhibitors and 5-HT1A partial agonists. Bioorganic Med. Chem. Lett. 2010, 20, 1114–1117. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Type of Fluorine | Fluorine Attached to an Aromatic Carbon | Fluorine Attached to Aliphatic Carbon |
|---|---|---|
| no. of positive fluorinated compounds ΔpPot > 0.3 | 556 | 21 |
| no. of negative fluorinated compounds ΔpPot < −0.3 | 619 | 97 |
| no. of positive AC | 16 | 1 |
| no. of negative AC | 22 | 15 |
| The Site of Fluorine Substitution | No. of Positive Fluorinated CPDs ΔpPot > 0.3 | No. of Negative Fluorinated CPDs ΔpPot < −0.3 |
|---|---|---|
| ortho | 54 | 21 |
| meta | 55 | 50 |
| para | 125 | 144 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietruś, W.; Kurczab, R.; Stumpfe, D.; Bojarski, A.J.; Bajorath, J. Data-Driven Analysis of Fluorination of Ligands of Aminergic G Protein Coupled Receptors. Biomolecules 2021, 11, 1647. https://doi.org/10.3390/biom11111647
Pietruś W, Kurczab R, Stumpfe D, Bojarski AJ, Bajorath J. Data-Driven Analysis of Fluorination of Ligands of Aminergic G Protein Coupled Receptors. Biomolecules. 2021; 11(11):1647. https://doi.org/10.3390/biom11111647
Chicago/Turabian StylePietruś, Wojciech, Rafał Kurczab, Dagmar Stumpfe, Andrzej J. Bojarski, and Jürgen Bajorath. 2021. "Data-Driven Analysis of Fluorination of Ligands of Aminergic G Protein Coupled Receptors" Biomolecules 11, no. 11: 1647. https://doi.org/10.3390/biom11111647