
Hyperhomocysteinemia: Metabolic Role and Animal Studies with a Focus on Cognitive Performance and Decline—A Review


Abstract
:1. Introduction
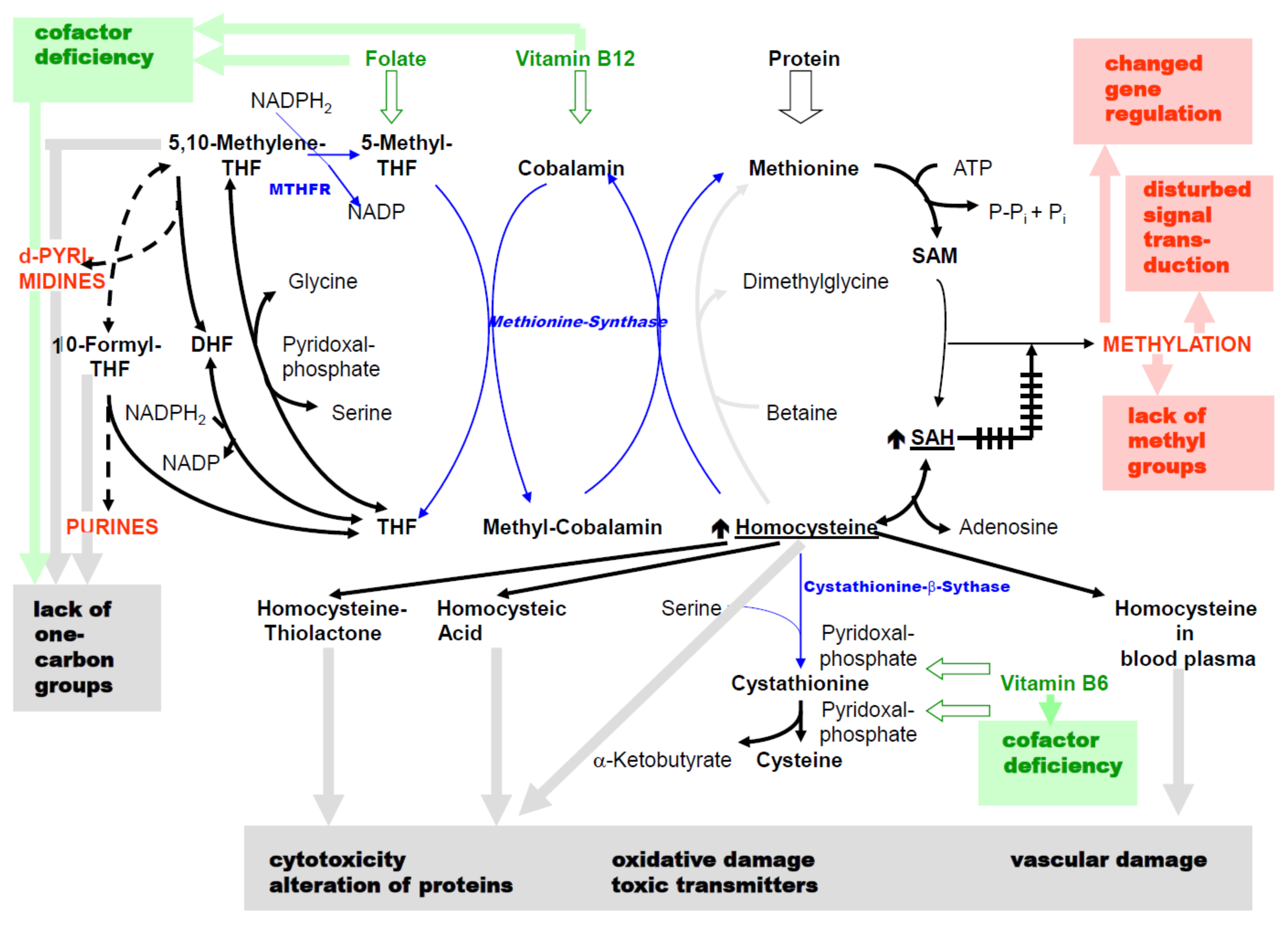
2. C1 Metabolism and HHCys
2.1. Reactions of the C1 Metabolism and Its Main Products
2.1.1. Thermodynamic Features
- (a)
- The reaction catalyzed by 5,10-methylenetetrahydrofolate reductase (MTHFR) proceeds almost completely unidirectional to 5-methyl-THF under normal metabolic conditions [5]. There is a reason for the so-called folic acid trap [6,7]: if there is a pronounced vitamin B12 deficiency, there is no re-methylation of HCys via methionine synthase (Figure 1). Even with sufficient folic acid intake, it accumulates as 5-methyl-THF potentially resulting in a deficiency in C1 compounds of the folic acid cycle.
- (b)
- The reaction of S-adenosyl homocysteine (SAH) to HCys (by the SAH hydrolase) tends towards SAH formation [8]. In this way, the cellular HCys concentration is kept low under normal metabolic conditions.
2.1.2. Special Features of the Enzyme Equipment and Kinetics
- (a)
- (b)
- The cellular concentrations of total HCys for most organs are 2–7 nmol/g wet weight [10]. Calculating with approximately 70% cell water results in concentrations of 3–10 µM. The KM values for HCys of the initiating enzymes of re-methylation (methionine synthase) and transsulfuration (cystathionine-β-synthase) are 0.06 mM and approximately 10 mM [11] and thus, differ by three orders of magnitude. Serine, the second substrate of cystathionine-β-synthase (CBS), also has a high KM value of 2 mM [12]. From this, it can be concluded that if there is an adequate supply with folic acid, vitamin B12 and B6, HCys is predominantly re-methylated.
- (c)
- Transsulfuration is not possible in some tissues, because there is no expression of CBS (heart, vessels, lungs, adrenal gland, spleen, testes) or cystathionase (brain, adipose tissue) [11].
- (d)
- The availability of sufficient SAM as a substrate for the majority of methylation reactions is a crucial function of C1 metabolism. In humans, 6–8 g SAM are synthesized daily [13]. Its synthesis is largely ensured by the effector functions of SAM and, at the same time, HCys metabolism is influenced, since SAM inhibits the MTHFR [14] and activates the CBS [15]. In cells that express both enzymes, when SAM levels rise (e.g., due to an abundant supply of methionine) this is irreversibly removed via transsulfuration. Owing to the high KM value of CBS (cf. above) enhanced flux rate through transsulfuration is accompanied by an increase in cellular HCys concentration. When there is a deficiency of SAM, re-methylation of HCys is stimulated.
- (e)
- SAH is a potent inhibitor of most SAM-dependent methylation reactions [16]. However, the consequences are different for individual methylations, as will be explained later.
- (f)
- A special kind of methylation cycle arises from the ability of the methionine synthase to catalyze also protein-bound HCys, as in the case of the D4 dopamine receptor (D4). Stimulation of D4-bound methionine leads via D4-bound SAM to methylation of membrane phospholipids [17].
2.2. Principal Causes of C1 Metabolic Disorders
2.3. HCys as a Diagnostic Measurable Biomarker of Disorders
- (a)
- Because HCys is transported out of the cells, the concentrations in the extracellular space and plasma do not have to correspond to those in the cells, which are responsible for the increased production. The liver is the main organ for HCys formation [39]. However, when HCys formation and export are stimulated, the cellular concentration in the liver remains relatively constant [10]. Cultured endothelial cells continuously export HCys into the medium and keep the cellular concentration at a significantly lower level [38]. In contrast, the addition of HCys to the medium (100 µM) leads to absorption and increases the intracellular concentration [38]. It can, therefore, be assumed that endothelial cells have only a small capacity to re-utilize HCys. Increase in HCys levels in plasma and extracellular space is not only effective at, but also in endothelial cells.
- (b)
- Only free HCys is reactive. The ratio of free to protein-bound HCys is different intracellularly than in blood plasma [10]. For example, approximately 4.5 and 3 nmol/g wet weight for free and bound HCys were measured in rat liver, which exchange with a half-life in the range of seconds. The quotient of free/bound HCys is 1.47 for rat liver. For cerebrum and cerebellum, it is 2.72 and 17.81, respectively. Free HCys is exported [10].
2.4. Principal Pathological Mechanisms with Morbid Effects in C1 Metabolic Disorders
- (a)
- The supply of C1 compounds from THF metabolites is reduced due to folic acid deficiency. This results in lack of nucleotides in energy metabolism and impairment of DNA and RNA synthesis. There is also impairment of mitotic rate.
- (b)
- In addition to reduced HCys transsulfuration, vitamin B6 deficiency causes inhibition of numerous pyridoxal phosphate-dependent reactions in amino acid metabolism.
- (c)
- Vitamin B12 deficiency leads to the accumulation of HCys.
- (a)
- The KM values for SAM and the KI values for SAH are different for individual methyltransferases and differ between the various enzymes by almost three orders of magnitude [52,53]. A changed SAM/SAH quotient can either do nothing at all, e.g., if the enzyme continues to work in the VMax range, or result in changes in methylation.
- (b)
- There are “buffer reactions” without metabolic effects, such as the methylation of glycine to sarcosine by glycine-N-methyltransferase, which regulates the SAM concentration [3].
- (c)
Experimental Use of Methionine or HCys
- (a)
- In humans, D- and DL-methionine show only 30% and 65% effectiveness, respectively, compared with L-methionine regarding the nitrogen balance [65].
- (b)
- In chicks, D-HCys is only re-methylated to methionine to about 25% via methionine synthase, compared with L-HCys [66].
- (c)
- In a methionine-deficient diet, L-HCys can replace 65% of the growth-promoting effect of L-methionine via this reaction, but D-HCys only 7% [67].
- (d)
2.5. Homocystinuria as a Result of an Existing Homozygous Defect in CBS—Witness of HCys Pathology
- (a)
- There are no other causes of disorders in C1 metabolism, such as a vitamin deficiency.
- (b)
- (c)
- CBS also catalyzes the formation of hydrogen sulfide (H2S), which may be diminished. There are, however, two further enzymes that catalyze H2S formation from cysteine: cystathionine-γ-lyase and 3-mercaptopyruvate sulfurtransferase [70]. Moreover, HCys was found to upregulate cystathionine-γ-lyase in cardiomyocytes and also in vivo (Cbs+/− mice), the enzyme was upregulated [71]. Furthermore, even in the absence of pyridoxal-5′-phosphate, brain homogenates of CBS-knockout mice produced H2S levels from cysteine similar to those of wild-type mice by 3-mercaptopyruvate sulfurtransferase in combination with cysteine aminotransferase [72].
3. Diseases in which C1 Metabolic Disturbances and HHCys Are Significantly Involved in the Pathogenesis
4. Animal Studies on HHCys—Literature Search Results and Discussion
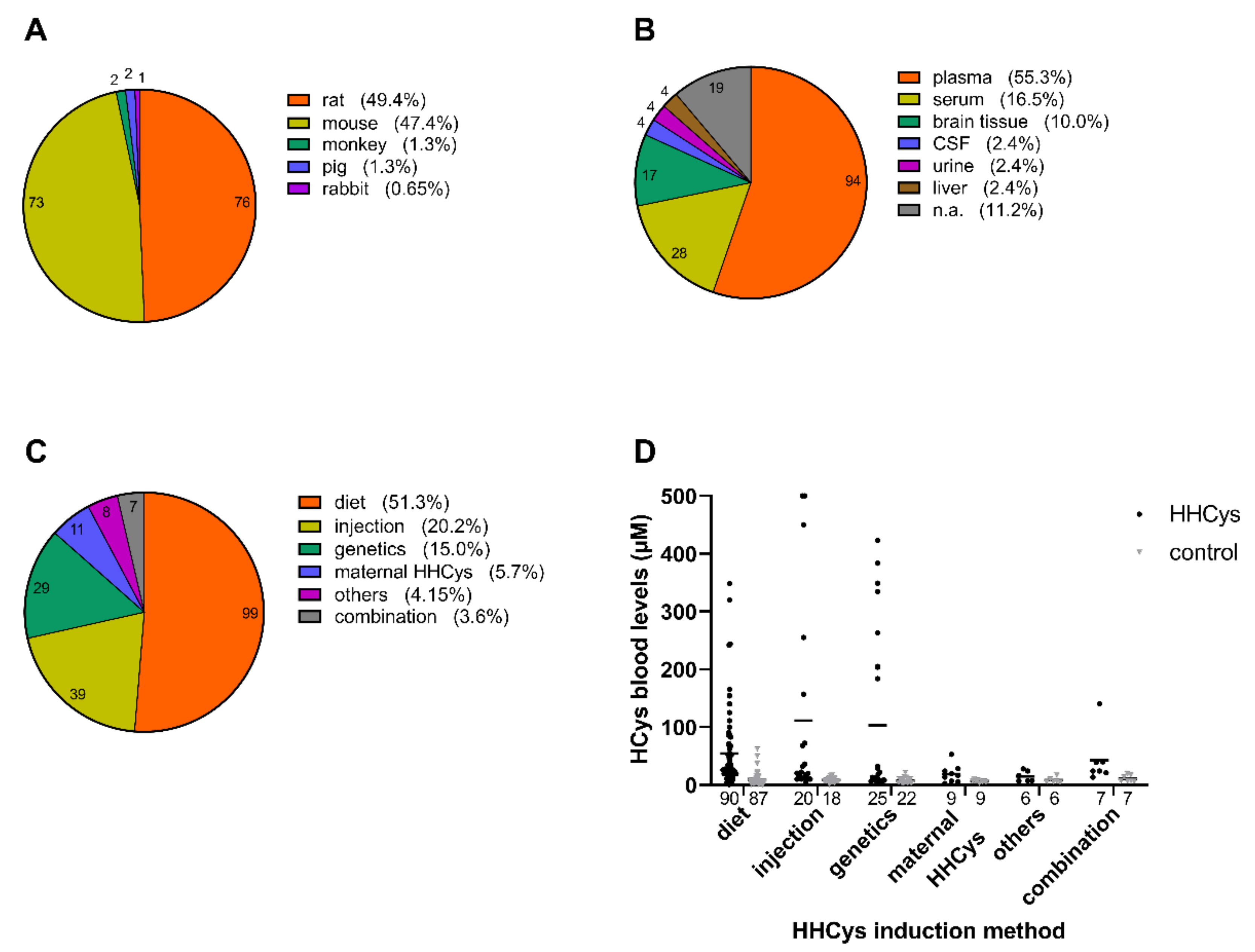
4.1. HHCys Induction Methods in Animal Models
4.1.1. Dietary Induction
4.1.2. Parenteral Induction
4.1.3. Genetic Induction
4.1.4. Impact of Maternal HHCys
4.1.5. Combinatory and Other Induction Methods
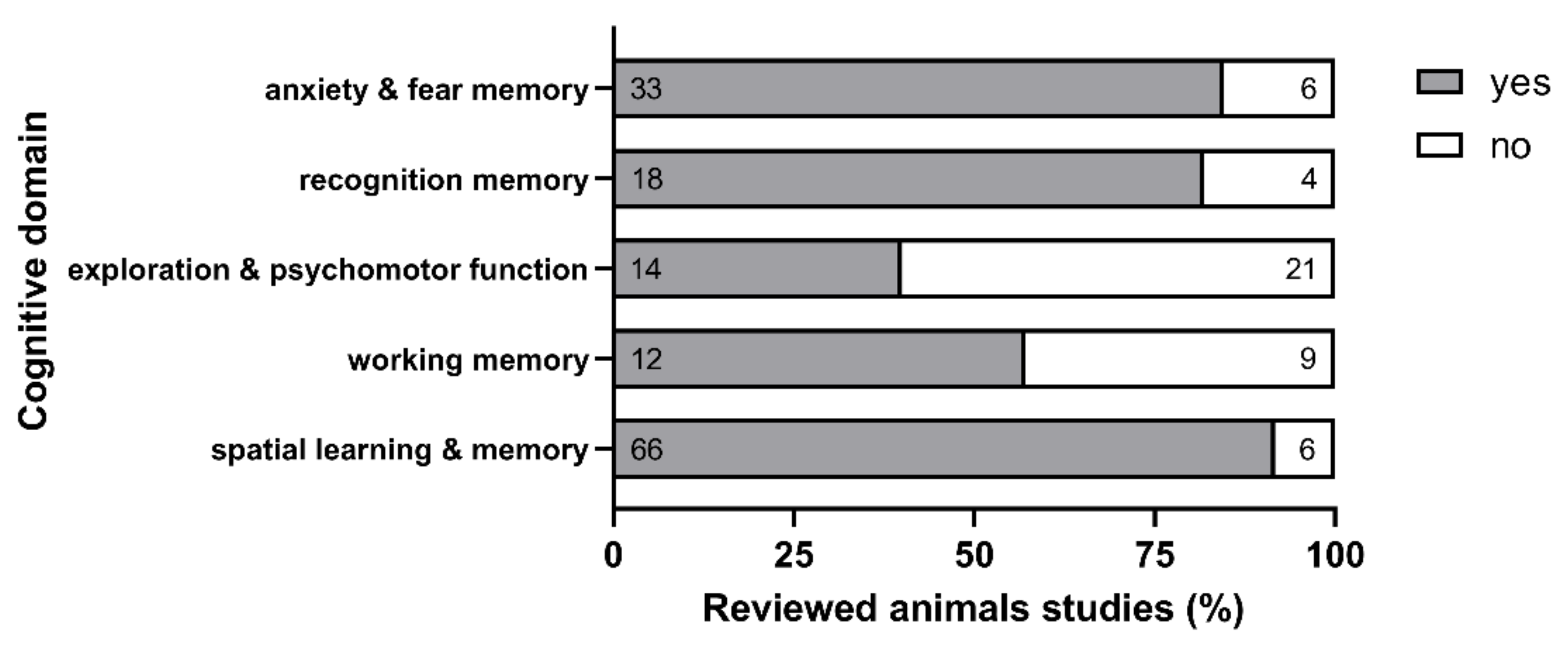
4.2. HHCys Impact on Cognition in Animal Models
5. Summary and Conclusion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease course over years or decades without symptoms. Elevated HCys values are pathogenetically involved. Clinical symptoms first become prevalent through complications of atheromatous plaques with a different pathogenesis, in which B-vitamins and HCys hardly play a role. Initial endothelial cell damage—Rev [61,161]; also see Figure 4A–C: Reduced formation and efficacy of NO → inadequate vasodilatation in response to atherosclerosis-promoting stimuli. After coronary angiographic localization of such functional restrictions, vascular constrictions can be found after years in patients with acute coronary syndrome [162]. HCys causes increased formation of ROS (see Figure 4E) → cell activation with increased formation of adhesion molecules and pro-inflammatory cytokines; cell damage; apoptosis. S-homocysteinylation of endothelial proteins → loss of function (see Figure 4D). N-homocysteinylation by HCys-thiolactone → cytotoxicity (see Figure 4A). With HCys, the cellular SAH concentration increases → altered DNA or RNA methylation (see Figure 3) → reduced expression of enzymes with an antioxidant effect. Decrease in the thromboresistance of the endothelial surface by promoting coagulation and inhibiting anticoagulant and fibrinolytic mechanisms. Plasma lipids and white blood cells—Rev [161]: Oxidation of LDL → uptake by white blood cells → promotion of foam cell formation. Increase in chemotactic motility of white blood cells. Smooth muscle cells—Rev [61]: Oxidative stress → activation of the transcription factor NFκB → proliferation. Platelets—Rev [61]: Increase in thromboxane A2 synthesis → promotion of reactivity and aggregation. | Studies on the influence of plasma HCys concentration on atherosclerosis consequences: Recording of the period until the onset of symptoms. Elimination of conventional risk factors. Meta-analysis from >70 case-control studies as well as prospective studies with >20,000 subjects: An increase in HCys of 5 µM results in a 33% increase in risk for ischemic heart disease and 59% for ischemic stroke [163]. Meta-analysis from 12 prospective studies with >9000 subjects: Lowering HCys by 3 μM results in an 11% decrease in risk for ischemic heart disease and 19% for ischemic stroke [164]. Peripheral occlusion: case-control studies, with significantly higher levels of HCys than controls [165]. Renal insufficiency: high HCys levels (cf. Section 2.3). Greatly increased risk for all consequences of atherosclerosis, which are the main causes of death [166]. Prospective intervention studies—Rev [167,168]: A total of 15 studies had the effect of at least one of vitamin B6, B12 or folate was compared with placebo. 11 of these studies were secondary preventive—after a clinical event such as myocardial infarction—the remaining 4 studies in renal insufficiency requiring dialysis → no primary prevention overall. Results: heterogeneous/controversial. For ischemic stroke only, 25% risk reduction with the three B-vitamins. Only one primary preventive intervention study: >20,000 subjects with hypertension received folic acid + ACE inhibitors (Enalapril) versus only ACE inhibitors for 5 years: 34% reduction in ischemic stroke, 20% reduction in the combination of stroke, myocardial infarction, cardiovascular death. Renal insufficiency: meta-analysis of a total of 3886 patients: Monotherapy with folic acid resulted in a significant reduction in cardiovascular endpoints by 15%; in patients without (additional) dietary folic acid fortification by 20% [169]. |
| With a comparable HCys level, oxidative stress is stronger than in controls [61], directly detectable in myocardial fibrils (bypass surgery): H2O2 production ↑, antioxidants ↓ [170]. Metformin therapy inhibits intestinal absorption of vitamin B12 and folic acid [171]. | Plasma HCys level as in controls, but higher if nephropathy is present. HCys is more strongly associated with atherosclerosis and its consequences than in non-diabetes controls [172]. Primarily preventive, folic acid supplementation lowers HCys and insulin levels, reduces insulin resistance, normalizes glucose homeostasis and atherogenic plasma lipid profile [173]. Metformin therapy lowers B12 levels, increases HCys levels and intensifies diabetic neuropathy [174]. |
| HCys causes a decrease in the thromboresistance of the endothelial surface by promoting coagulation and inhibiting anticoagulatory and fibrinolytic mechanisms [61]. | Meta-analyses of clinical studies—[175,176]: An increase in HCys of 5 μM increases the risk of deep vein thrombosis by 60% (case-control studies) or 27% (prospective studies). Patients with deep vein thrombosis and pulmonary embolism have significantly reduced plasma folic acid and/or vitamin B12 levels. Intervention studies so far unsatisfactory. The combination of HHCys and factor V (Leiden) is multiplicative [177]. |
| Reduced transmitter formation—Rev [52]; also see Figure 3: Folic acid and SAM necessary for the synthesis of serotonin, noradrenaline and dopamine, both directly as well as via the synthesis of tetrahydrobiopterin. Significant changes in patients with depression: increase in HCys (plasma), decrease in folic acid (plasma, erythrocytes, liquor), SAM (liquor) and metabolites of the 3 transmitters (liquor) | Case control studies: Plasma HCys >10 μM → doubling the risk of depression [178]. Intake of vitamin B6, B12, folate correlates negatively with the occurrence of depression (12 years observation period) [179]. Intervention studies: The three B-vitamins versus placebo in patients at risk of depression for 7 years: significantly lower frequency [180]. Therapy with antidepressants in combination with folic acid, 5-methyl-tetrahydrofolate or SAM: better effect than antidepressants alone [52]. |
| HCys and oxidation products (HCA) activate NMDA receptors → excitotoxicity (cellular Ca2+ increase → activation of proteases and radical formation → cell death = neuron degeneration)—see Figure 4F | Case-control studies: significant deviations in plasma levels in autistic children: HCys ↑; vitamin B6, B12, folate ↓. Interventional study: folic acid supplementation lowers plasma HCys levels and reduces deficits in cognition, communication and social behavior. |
| Levodopa is degraded by methylation → significantly higher HCys levels than in untreated Parkinson’s patients → increased risk of stroke, coronary artery disease, dementia and peripheral neuropathy [181]. Anticonvulsants, especially valproate, influence the metabolism of folic acid (inhibition of cellular receptors), vitamin B6 (increased degradation) and reduce betaine uptake → lowering of plasma level of the two vitamins and increase in HCys [182]—see Figure 2 Particular risk: carrier of the TT variant of the C677T mutation of the MTHFR—see Table 1 | Substitution with vitamin B6, B12, folate lowers HCys levels [183]. Significantly more femoral neck and vertebral fractures [184] and brain atrophy Rev [185]. Pregnancy: 10-fold increased risk of abortion, malformations in 6–11% of newborns, often cognitive deficits [186]. Improvement after supplementation with folic acid and B6 [184] or folic acid and B12 [186]. |
| Most frequent cause: damage to the peripheral myelin protein-22 [187]. HHCys → methylation disorders (see Figure 3): Hypo-methylation of Arg107 of the basic myelin protein → loss of binding for acidic lipids → disrupted lamellar formation. | HCys ↑ causally affects neuropathies in: Parkinson’s disease under levodopa therapy, type 2 diabetes mellitus (especially with metformin therapy), chronic alcoholism. Supplementation with vitamin B6, B12, folate improves the symptoms [170]. |
| Pregnancy changes laboratory parameters for assessing C1 metabolism: U-shaped course with a lowering of vitamin B6, B12, folate and HCys in plasma in early pregnancy. Only holotranscobalamin remains constant [188]; only HCys levels in the interval are meaningful. Threshold for women of childbearing age: 9 μM | Evaluation of >14,000 pregnancies: HCys level in the interval >8.9 μM → significantly increased risk of preeclampsia, premature birth, stillbirth, low birth weight, neural tube defects [189]. |
| |
|
|
| |
|
|
| |
|
|
| Plasma concentrations of vitamin B6, B12, folate and HCys are similar to those in seminal fluid. Every alteration of C1 metabolism associated with HCys ↑ leads to DNA fragmentation, telomere shortening, a different methylation pattern (see Figure 3) and radical formation (see Figure 4E) in sperm and oocytes. Hypomethylation of IGF2_H19 locus in men correlates with infertility. In vitro fertilization—quality of the embryo: Positive correlation with B12 content and negative correlation with HCys concentration in plasma and follicular fluid | Prospective study—approx. 20,000 women, 8 years: Infertility correlates negatively with daily folic acid intake. MTHFR C677T-TT carriers: more often infertile. Significantly higher levels of HCys in spermatozoa in infertile men. In vitro fertilization—intervention: Supplementation with the three B-vitamins reduces DNA fragmentation in sperm, doubles the pregnancy rate and triples the birth rate. |
| Human retinal cell culture: HCys induces production of VEGF (vascular endothelial growth factor). Plasma HCys correlates with VEGF-concentration in vitreous humor | Direct correlation between plasma HCys level and risk of macular degeneration. Significantly higher plasma HCys levels in exudative macular degeneration than in dry macular degeneration and controls. Intervention study: three B-vitamins versus placebo for 7 years in 5000 subjects: 34% less macular degeneration. Diabetes mellitus: in patients significantly higher HCys levels in serum, vitreous humor and retina. |
| S-homocysteinylation (see Figure 4D) of collagen fibrils hinders the regular formation of the bone matrix → increased fragility with mostly unchanged bone density. | Prospective studies: on average about twice the risk of femoral neck fractures with plasma HCys ≥15 μM. MTHFR C677T-TT carrier: significantly increased fracture rate. Interventional studies—three B-vitamins versus placebo: negative if related to bone density and plasma bone turnover parameters; mostly positive when it comes to fracture rates. |
| Chronic stress: (1) Increased formation of ROS (see Figure 4E) → peroxynitrite anion ↑ in the respiratory chain → irreversible inhibition of cytochrome C oxidase → cellular energy production ↓. (2) Leukocytes from patients with fibromyalgia: hypo-methylation and increased mRNA formation (see Figure 3) of genes with sensory, adrenergic and immunological functions. Cobalamin acts as an intracellular antioxidant in high concentrations. | Plasma vitamin B12 and HCys levels correlate positively/negatively, with exhaustion, comprehensive psychopathological rating scale, pain and memory ability. Therapy with high doses of vitamin B12 (1–2 mg/day) and folic acid (1–5 mg/day). |
Appendix B
Appendix B.1. Literature Search Strategy
Appendix B.2. Literature Analysis
| Strategy to Induce HHCys | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet/Drinking Water | Injection | Genetic Manipulation | Maternal HHCys Impact | Others | Investigated Biological Matrix | Impact on Cognitive Performance | |||||||||||||||
| Publication | Animal Species | B-vit. def. | Met suppl. | HCys suppl. | Others | HCys | Others | CBS | MTHFR | Others | Blood Levels (µM): ↑HCys vs. Control/Baseline Data (Where Applicable) | Plasma | Serum | Brain Tissue | CSF | Urine | Liver Tissue | Cognitive Domain & Reported Effects of HHCys yes (+) or no (-) | Investigation of Potential Treatment Option | ||
| [83] | rat | 28.8 vs. 6.3 | spatial learning & memory (+) | ozagrel | |||||||||||||||||
| [215] | rat | 2.3 vs. 0.9 1 | spatial learning & memory (+) | edaravone | |||||||||||||||||
| [80] | mouse | 348.2 vs. 7.7 2;3;4 | exploration (-); anxiety (-); spatial learning & memory (-) ; recognition memory (-); others (-) | B-vitamins, PUFA, Fortasyn® Connect-like diet | |||||||||||||||||
| [216] | rat | 5.65 vs. 4.85 (offspring) | offspring: working memory (+) | mild transient neonatal hypoxia | |||||||||||||||||
| [217] | mouse | 19.0 vs. < 5 (WT); 14.7 vs. < 5 (KI) | n.a. | n.a. | |||||||||||||||||
| [218] | rat | 11.22 vs. 7.08 | n.a. | n.a. | |||||||||||||||||
| [104] | rat | 27.3 vs. 7.9 (dams); 19.5 vs. 6.3 (offspring) | offspring: exploration (+); anxiety (+); psychomotor function (+); working memory (+); spatial learning & memory (+); others (+) | sodium hydrosulfide | |||||||||||||||||
| [219] | rat | n.a. | exploration (-); spatial learning & memory (+); fear memory (+) | synthetic tricyclic sulfonamide PP2A activators | |||||||||||||||||
| [220] | mouse | n.a. | spatial learning & memory (+) | maternal choline supplementation | |||||||||||||||||
| [123] | rat | 10.1 vs. 6.1 | exploration (-); recognition memory (+); spatial learning & memory (+) | emodin | |||||||||||||||||
| [221] | rat | 11.38 vs. 7.15 | n.a. | n.a. | |||||||||||||||||
| [222] | mouse | 71.5 vs. 4.9 | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [223] | mouse | 423 vs. < 16 | anxiety (n.a.); exploration (n.a.); others (+) | methionine restriction, enzyme replacement | |||||||||||||||||
| [224] | mouse | 140.50 vs. < 5 | spatial learning & memory (+); others (-) | n.a. | |||||||||||||||||
| [225] | mouse | 22 vs. 17 (injection); 24 vs. 17 (age) 1 | recognition memory (-); fear memory (+); spatial learning & memory (+) | B-vitamins, SAM | |||||||||||||||||
| [101] | mouse | 263 vs. 13 (CBS); 184 vs. 13 (CTH) | n.a. | n.a. | |||||||||||||||||
| [226] | rat | 13.13 vs. 8.5 | spatial learning & memory (-); recognition memory (-); anxiety (-) | betaine | |||||||||||||||||
| [227] | mouse | 82.93 vs. 5.89 (WT); 84.67 vs. 6.34 (KO) | n.a. | n.a. | |||||||||||||||||
| [105] | mouse | n.a. | exploration (+); anxiety (+); recognition memory (+); spatial learning & memory (+) | methionine restriction | |||||||||||||||||
| [122] | rat | 20 vs. 9 1 | spatial learning & memory (+) | liraglutide | |||||||||||||||||
| [228] | mouse | 13.97 vs. 8.55 (genetic); 18.93 vs. 8.55 (diet, WT); 38.87 vs. 13.97 (diet, Tg) | recognition memory (+); working memory (-); exploration (-); anxiety (+) | n.a. | |||||||||||||||||
| [229] | rat | 24 vs. 8 1 (offspring) | offspring: sensorimotor function (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [230] | rat | 17.5 vs. 8 | n.a. | B-vitamins | |||||||||||||||||
| [231] | rat | 22 vs. 8 (Met suppl.); 62 vs. 8 (B-vit. def. + Met suppl.) | exploration (+); anxiety (+) | statins | |||||||||||||||||
| [232] | mouse | n.a. | working memory (-); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [233] | rat | n.a. | spatial learning & memory (+) | Moringa oleifera extract | |||||||||||||||||
| [113] | rat | 28 vs. 10 1 | n.a. | epigallocatechin-3-gallate | |||||||||||||||||
| [90] | rat | 255.15 vs. 7.15 (acute); 16.64 vs. 7.15 (chronic) | n.a. | n.a. | |||||||||||||||||
| [234] | mouse | 52 vs. 22 1 | recognition memory (+) | n.a. | |||||||||||||||||
| [235] | rat | 0.59 vs. 0.3 1 | spatial learning & memory (+) | caffeine | |||||||||||||||||
| [236] | mouse | 22.01 vs. 14.43 | anxiety (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [237] | rat | 22 vs. 10 1 (dams) | offspring: sensorimotor function (+); spatial learning & memory (+) | folate | |||||||||||||||||
| [238] | rat | n.a. | exploration (-); spatial learning & memory (+); fear memory (+) | Ginkgo biloba extract | |||||||||||||||||
| [239] | rat | n.a. | working memory (+); anxiety (+) | hydrogen sulfide | |||||||||||||||||
| [118] | rat | n.a. 2 | exploration (-); anxiety (-); spatial learning & memory (+); recognition memory (+) | n.a. | |||||||||||||||||
| [111] | rat | 9 vs. 4.5 1 | working memory (+) | Vitis vinifera leaves polyphenols | |||||||||||||||||
| [102] | mouse | 29 vs. 10 (homozygous); 11 vs. 10 (heterozygous) 1 | spatial learning & memory (+); working memory (+); psychomotor function (-) | n.a. | |||||||||||||||||
| [240] | rat | n.a. | recognition memory (+); fear memory (+) | creatine | |||||||||||||||||
| [241] | rat | 36 vs. 4 1 | spatial learning & memory (+); anxiety (+); exploration (-); psychomotor function (-) | hydrogen sulfide | |||||||||||||||||
| [115] | rat | 22 vs. 7 (diet); 12 vs. 7 (injection); 24 vs. 7 (diet + injection) 1 | spatial learning & memory (+); recognition memory (+) | bosentan | |||||||||||||||||
| [242] | mouse | n.a. | working memory (-); fear memory (-) | genetic absence of ALOX5 | |||||||||||||||||
| [243] | mouse | n.a. | working memory (+); fear memory (+); spatial learning & memory (+) | ALOX5 inhibition (zileuton) | |||||||||||||||||
| [244] | rat | 153.79 vs. 62.21 3 | working memory (+); spatial learning & memory (+) | fisetin | |||||||||||||||||
| [245] | rat | 165.48 vs. 49.64 3 | working memory (+); spatial learning & memory (+) | hesperidin | |||||||||||||||||
| [246] | rat | n.a. | spatial learning & memory (+); recognition memory (+) | hydrogen sulfide | |||||||||||||||||
| [247] | mouse | 67.40 vs. < detection range (WT); 70.29 vs. < detection range (Tg) | spatial learning & memory (+) | anti-Aβ immunotherapy | |||||||||||||||||
| [106] | rat | 8.18 vs. 4.43 (diet); 7.37 vs. 4.43 (stress) | exploration (+); recognition memory (+); fear memory (+); spatial learning & memory (+) | B-vitamins, betaine | |||||||||||||||||
| [248] | rat | 26 vs. 15 (dams); 53 vs. 7 (offspring) 1 | n.a. | maternal vitamin B6 supplementation | |||||||||||||||||
| [249] | mouse | n.a. | recognition memory (+); fear memory (+) | hydrogen sulfide | |||||||||||||||||
| [250] | mouse | n.a. | fear memory (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [251] | mouse | n.a. | spatial learning & memory (+); working memory (+); recognition memory (+) | cinnamon | |||||||||||||||||
| [252] | mouse | 46.1 vs. 4.6 | spatial learning & memory (+) | Brazilian propolis extract | |||||||||||||||||
| [253] | mouse | n.a. 5 | working memory (+); fear memory (+) | betaine | |||||||||||||||||
| [254] | mouse | 22 vs. 14 (dams) 1 ; 28.4 vs. 9.8 (offspring) | offspring: recognition memory (+); working memory (-) | n.a. | |||||||||||||||||
| [110] | rat | n.a. 5 | exploration (+); others (+) | n.a. | |||||||||||||||||
| [255] | rat | n.a. | spatial learning & memory (+) | atractylenolide III | |||||||||||||||||
| [256] | mouse | 18 vs. 13 (WT); 26 vs. 14 (Tg) 1 | spatial learning & memory (+); psychomotor function (-); anxiety (+) | n.a. | |||||||||||||||||
| [257] | rat | 16.7 vs. 16.3 | n.a. | zinc | |||||||||||||||||
| [258] | rat | n.a. | fear memory (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [114] | rat | 16 vs. 7 1 | spatial learning & memory (+); fear memory (+) | fatty acids | |||||||||||||||||
| [259] | rat | n.a. | spatial learning & memory (+) | combination: acetylcholinesterase inhibitor + calcium channel blocker | |||||||||||||||||
| [260] | rat | n.a. | offspring: exploration (+); anxiety (+); fear memory (+) | n.a. | |||||||||||||||||
| [261] | mouse | 22 vs. 6 1 | locomotion (-); recognition memory (-) | n.a. | |||||||||||||||||
| [262] | mouse | n.a. | recognition memory (+) | ablation of MMP9 gene | |||||||||||||||||
| [103] | mouse | 13 vs. 3 (homozygous); 5 vs. 3 (heterozygous) 1 | recognition memory (+); working memory (+) | n.a. | |||||||||||||||||
| [263] | rat | n.a. | spatial learning & memory (+); recognition memory (+) | n.a. | |||||||||||||||||
| [81] | rat | 48 vs. 7 1 | exploration (-); anxiety (+); others (-) | n.a. | |||||||||||||||||
| [264] | mouse | n.a. | working memory (-); fear memory (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [265] | rat | 10 vs. 6 1 | spatial learning & memory (+) | hydroxysafflor yellow A | |||||||||||||||||
| [94] | rat | n.a. 2 | recognition memory (+) | memantine | |||||||||||||||||
| [112] | rat | 9.2 vs. 3.8 | spatial learning & memory (+) | rivastigmine (liposomal) | |||||||||||||||||
| [97] | mouse | 7.5 vs. 5.5 (age); 11 vs. 5.5 (genetic, adult); 13.5 vs. 7.5 (genetic, old) 1 | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [266] | mouse | 26 vs. 8 (WT); 54 vs. 9 (Tg) 1 | n.a. | n.a. | |||||||||||||||||
| [267] | mouse | 82.93 vs. 5.89 | spatial learning & memory (+); psychomotor function (-) | n.a. | |||||||||||||||||
| [268] | rat | n.a. | spatial learning & memory (+) | betaine | |||||||||||||||||
| [269] | rat | 19.16 vs. 5.21 | spatial learning & memory (+) | resveratrol | |||||||||||||||||
| [270] | rat | n.a. | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [98] | mouse | n.a. | psychomotor function (+); exploration (+); anxiety (+); recognition memory (+); working memory (+) | n.a. | |||||||||||||||||
| [271] | rat | 21.2 vs. 6.16 | spatial learning & memory (+) | diethyl dithio carbamate trihydrate, folacin | |||||||||||||||||
| [272] | rat | 21 vs. 7.4 (dams) | offspring: spatial learning & memory (+) | ginkgo biloba extract | |||||||||||||||||
| [273] | rat | 5.1 vs. 3.2 1 | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [274] | rat | 52.3 vs. 6.96 | exploration (+); anxiety (+); others (+) | n.a. | |||||||||||||||||
| [275] | mouse | 90.68 vs. 2.04 (WT); 118.75 vs. 0.41 (Tg) | fear memory (-); spatial learning & memory (-) | SAM | |||||||||||||||||
| [276] | rat | 21.2 vs. 6.16 | spatial learning & memory (+) | pioglitazone; rosiglitazone | |||||||||||||||||
| [277] | mouse | 100 vs. 8 (Met suppl.); 70 vs. 8 (B-vit. def.) 1 | working memory (-); fear memory (-) | n.a. | |||||||||||||||||
| [278] | rat | n.a. | spatial learning & memory (+); fear memory (+) | acetyl-L-carnitine | |||||||||||||||||
| [279] | rat | n.a. 2 | recognition memory (+); spatial learning & memory (+) | dextromethorphan | |||||||||||||||||
| [78] | mouse | 111 vs. 5 (WT); 76.4 vs. 3.8 (Tg) | n.a. | SAM | |||||||||||||||||
| [280] | pig | 6.88 vs. 5.45 | exploration (+); psychomotor function (-); working memory (-); others (+) | folate | |||||||||||||||||
| [109] | rat | n.a. | n.a. | N-acetyl cysteine + α-lipoic acid + α-tocopherol | |||||||||||||||||
| [281] | rat | n.a. | fear memory (+); exploration (-) | curcumin | |||||||||||||||||
| [282] | mouse | n.a. | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [210] | rat | ∼500 µM vs. n.a. | n.a. | n.a. | |||||||||||||||||
| [283] | mouse | 2.39 vs. 2.37 (offspring) | offspring: exploration (-); anxiety (-) | n.a. | |||||||||||||||||
| [87] | rat | 26.7 vs. 10.4 | spatial learning & memory (+) | n.a. | |||||||||||||||||
| [130] | mouse | n.a. 2 | spatial learning & memory (+) | anti-HCA antibody | |||||||||||||||||
| [213] | mouse | 16.8 vs. 3.4 | fear memory (+) | n.a. | |||||||||||||||||
| [284] | mouse | 155 vs. 5 1 | n.a. | n.a. | |||||||||||||||||
| [285] | mouse | 30 vs. 6 1 | n.a. | n.a. | |||||||||||||||||
| [286] | mouse | 35.4 vs. 6.33 | others (+) | n.a. | |||||||||||||||||
| [287] | rat | n.a. | psychomotor function (-); fear memory (+) | folate | |||||||||||||||||
| [288] | rat | 16.5 vs. 6.8 (offspring) | offspring: sensorimotor function (+); spatial learning & memory (+); others (-) | short-term neonatal hypoxia | |||||||||||||||||
| [289] | rat | 10.2 vs. 6.2 | spatial learning & memory (+) | B-vitamins | |||||||||||||||||
| [92] | mouse | 32.1 vs. 11.6 | n.a. | n.a. | |||||||||||||||||
| [290] | mouse | 67 vs. 8.5 (WT); 49.9 vs. 9.6 (Tg) | exploration (+); spatial learning & memory (+); anxiety (+); others (+) | n.a. | |||||||||||||||||
| [96] | mouse | 257–365 vs. 15.4–25.4 (diff. strains) | exploration (-); anxiety (-); fear memory (+) | n.a. | |||||||||||||||||
| [291] | rat | 24.8 vs. 6.8 (dams) | offspring: spatial learning & memory (+) | melatonin | |||||||||||||||||
| [292] | rat | 31.3 vs. 4.2 (B-vit. def.); 31.2 vs. 4.2 (B-vit. def. + Met suppl.) | spatial learning & memory (+); psychomotor function (-) | methionine | |||||||||||||||||
| [293] | mouse | 28.7 vs. 5.2 (B-vit. def.); 13.9 vs. 5.2 (Met suppl.) | spatial learning & memory (+); psychomotor function (-) | n.a. | |||||||||||||||||
| [79] | mouse | 320 vs. 0.2 (WT); 450 vs. 1 (Tg) 1 | spatial learning & memory (-) | n.a. | |||||||||||||||||
| [294] | mouse | 7.3 vs. 4.0 | exploration (+); anxiety (+); working memory (-); psychomotor function (+); spatial learning & memory (-) | n.a. | |||||||||||||||||
| [295] | rat | 10.2 vs. 6.2 1 | n.a. | B-vitamins | |||||||||||||||||
| [296] | rat | 26 vs. 6 (dams) | offspring: spatial learning & memory (+) | n.a. | |||||||||||||||||
| [203] | rat | 13.3 vs. 6.8 (offspring) | offspring: sensorimotor function (-); anxiety (+); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [212] | mouse | 101 vs. 37 (WT); 178 vs. 103 (Tg) | working memory (-); spatial learning & memory (+) | n.a. | |||||||||||||||||
| [84] | mouse | 243.7 vs. 5.1 (B-vit. def.); 86.7 vs. 5.1 (B-vit. def. + Met suppl.) | spatial learning & memory (+); psychomotor function (-); exploration (-) | B-vitamins | |||||||||||||||||
| [297] | mouse | 12.6 vs. 7.9 | n.a. | n.a. | |||||||||||||||||
| [211] | rat | 4.5 vs. 2.9 1 | spatial learning & memory (-) | n.a. | |||||||||||||||||
| [298] | rat | 20 vs. 7.5 1 | fear memory (+); spatial learning & memory (+) | melatonin | |||||||||||||||||
| [299] | mouse | 205 vs. 3.9 | n.a. | n.a. | |||||||||||||||||
| [300] | rat | 26.2 vs. 6.5 | n.a. | folate | |||||||||||||||||
| [301] | rat | 400–500 vs. 10 | spatial learning & memory (+); working memory (+); exploration (-) | n.a. | |||||||||||||||||
| [302] | mouse | 25 vs. 2 (WT); 27 vs. 3 (Tg) 1 | n.a. | n.a. | |||||||||||||||||
| [303] | mouse | 5.3 vs. 3.25 (heterozygous); 32.3 vs. 3.25 (homozygous) | n.a. | n.a. | |||||||||||||||||
| [304] | mouse | 125 vs. 9 | others (+) | n.a. | |||||||||||||||||
| Strategy to Induce HHCys | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet/Drinking Water | Injection | Genetic Manipulation | Maternal HHCys Impact | Others | Investigated Biological Matrix | ||||||||||||||
| Publication | Animal Species | B-vit. def. | Met suppl. | HCys suppl. | Others | HCys | Others | CBS | MTHFR | Others | Blood Levels (µM): ↑HCys vs. Control/Baseline Data (Where Applicable) | Plasma | Serum | Brain Tissue | CSF | Urine | Liver Tissue | ||
| [305] | mouse | 6.5 vs. 5.1 1 (offspring) | |||||||||||||||||
| [85] | mouse | 243.7 vs. 4.6 (B-vit. def.); 86 vs. 4.6 (B-vit. def. + Met suppl.) | |||||||||||||||||
| [306] | mouse | 349 vs. n.a. | |||||||||||||||||
| [307] | pig | 72.33 vs. 10.53 | |||||||||||||||||
| [308] | rat | 34.1 vs. 15.1 | |||||||||||||||||
| [309] | mouse | 383.6 vs. n.a. | |||||||||||||||||
| [310] | mouse | 19 vs. 10 (genetic); 16 vs. 10 (diet, WT);40 vs. 19 (diet, Tg) 1 | |||||||||||||||||
| [311] | rat | 45 vs. 15 (Met suppl.); 65 vs. 15 (GAA) 1 | |||||||||||||||||
| [312] | mouse | 9 vs. 1.5 1 | |||||||||||||||||
| [313] | mouse | 51.8 vs. 3.0 (Met suppl.); 21.4 vs. 3.0 (HCys suppl.) | |||||||||||||||||
| [95] | rat | 140 vs. 20 (diet); 68 vs. 15 (injection) 1 | |||||||||||||||||
| [314] | mouse | 3.8 vs. 3.7 (genetic); 40.7 vs. 3.7 (diet, WT); 140.3 vs. 6.8 (diet, Tg) | |||||||||||||||||
| [315] | mouse | 23.5 vs. 4.1 | |||||||||||||||||
| [209] | mouse | 4.0 vs. 3.38 | |||||||||||||||||
| [86] | mouse | 4.5 vs. 3 (genetic); 4.4 vs. 3 (Met suppl., WT); 8.4 vs. 3 (B-vit. def., WT); 9.5 vs. 3 (Met suppl. + B-vit. def., WT) 1 | |||||||||||||||||
| [316] | rabbit | 20.3 vs. 12.3 | |||||||||||||||||
| [317] | mouse | 242 vs. 13 | |||||||||||||||||
| [318] | mouse | 8.2 vs. 4.0 | |||||||||||||||||
| [319] | mouse | 53.6 vs. 9.46 (Met suppl.); 51.4 vs. 9.46 (HCys suppl.) | |||||||||||||||||
| [107] | mouse | 24.5 vs. 2.6 | |||||||||||||||||
| [320] | rat | 19.5 vs. 6.15 | |||||||||||||||||
| [321] | rat | 500 vs. n.a. | |||||||||||||||||
| [322] | mouse | 8.3 vs. 5.0 (genetic); 17.2 vs. 5.0 diet, WT); 21.2 vs. 17.2 (diet, Tg) | |||||||||||||||||
| [323] | mouse | 6.3 vs. 4.1 (genetic); 13.0 vs. 4.1 (diet, WT); 23.9 vs. 6.3 (diet, Tg) | |||||||||||||||||
| [88] | rat | 15.5 vs. 10.5 1 | |||||||||||||||||
| [89] | rat | 23.6 vs. 11.0 | |||||||||||||||||
| [324] | monkey | 10.6 vs. 4.0 | |||||||||||||||||
| [325] | mouse | 13.5 vs. 6.1 (heterozygous); 203.6 vs. 6.1 (homozygous) | |||||||||||||||||
| [326] | monkey | 157 vs. 1 | |||||||||||||||||
References
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Wei, X.; Jiang, D.-S. Protein methylation functions as the posttranslational modification switch to regulate autophagy. Cell. Mol. Life Sci. 2019, 76, 3711–3722. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-Methyltransferase and Regulation of S-Adenosylmethionine Levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrossian, T.C.; Clarke, S.G. Uncovering the Human Methyltransferasome. Mol. Cell. Proteom. 2011, 10, M110.000976. [Google Scholar] [CrossRef] [Green Version]
- Katzen, H.M.; Buchanan, J.M. Enzymatic Synthesis of the Methyl Group of Methionine. J. Biol. Chem. 1965, 240, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Selhub, J. Homocysteine Metabolism. Ann. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [Green Version]
- Scott, J. The Methyl Folate Trap—A physiological response in man to prevent methyl group deficiency in kwashiorkor (methionine deficiency) and an explanation for folic-acid-induced exacerbation of subacute combined degeneration in pernicious anaemia. Lancet 1981, 318, 337–340. [Google Scholar] [CrossRef]
- De la Haba, G.; Cantoni, G.L. The Enzymatic Synthesis of S-Adenosyl-l-homocysteine from Adenosine and Homocysteine. J. Biol. Chem. 1959, 234, 603–608. [Google Scholar] [CrossRef]
- Sunden, S.L.F.; Renduchintala, M.S.; Park, E.I.; Miklasz, S.D.; Garrow, T.A. Betaine–Homocysteine Methyltransferase Expression in Porcine and Human Tissues and Chromosomal Localization of the Human Gene. Arch. Biochem. Biophys. 1997, 345, 171–174. [Google Scholar] [CrossRef]
- Svardal, A.; Refsum, H.; Ueland, P.M. Determination of in vivo protein binding of homocysteine and its relation to free homocysteine in the liver and other tissues of the rat. J. Biol. Chem. 1986, 261, 3156–3163. [Google Scholar] [CrossRef]
- Finkelstein, J.D. The Regulation of Homocysteine Metabolism. In Homocysteine Metabolism: From Basic Science to Clinical Medicine. Developments in Cardiovascular Medicine; Graham, I., Refsum, H., Rosenberg, I.H., Ueland, P.M., Shuman, J.M., Eds.; Springer: Boston, MA, USA, 1997; Volume 196, pp. 3–9. [Google Scholar]
- Kery, V.; Poneleit, L.; Kraus, J.P. Trypsin Cleavage of Human Cystathionine β-Synthase into an Evolutionarily Conserved Active Core: Structural and Functional Consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef]
- Matsubara, K.; Neafsey, E.J.; Collins, M.A. Novel S-Adenosylmethionine-Dependent Indole-N-Methylation of Beta-Carbolines in Brain Particulate Fractions. J. Neurochem. 1992, 59, 511–518. [Google Scholar] [CrossRef]
- Jencks, D.A.; Mathews, R.G. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J. Biol. Chem. 1987, 262, 2485–2493. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Kyle, W.E.; Martin, J.J.; Pick, A.-M. Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem. Biophys. Res. Commun. 1975, 66, 81–87. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S -adenosylmethionine, S -adenosylhomocysteine, and transmethylation reactions. Can. J. Biochem. 1979, 57, 56–64. [Google Scholar] [CrossRef]
- Hodgson, N.W.; Waly, M.I.; Trivedi, M.S.; Power-Charnitsky, V.-A.; Deth, R.C. Methylation-related metabolic effects of D4 dopamine receptor expression and activation. Transl. Psychiatry 2019, 9, 295. [Google Scholar] [CrossRef]
- Herrmann, W.; Obeid, R. Cobalamin Deficiency. In Water Soluble Vitamins. Subcellular Biochemistry; Stanger, O., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 56, pp. 301–322. [Google Scholar]
- Javid, P.; Christensen, E. Vegetarians are at high risk of vitamin B12 deficiency. Ugeskr. Laeger 2016, 178, V06150484. [Google Scholar]
- Andres, E. Vitamin B12 (cobalamin) deficiency in elderly patients. Can. Med. Assoc. J. 2004, 171, 251–259. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Vitamin and Mineral Requirements in Human Nutrition, 2nd ed.; Report of a joint FAO/WHO expert consultation; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Martiniak, Y.; Heuer, T.; Hoffmann, I. Intake of dietary folate and folic acid in Germany based on different scenarios for food fortification with folic acid. Eur. J. Nutr. 2015, 54, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Field, M.S.; Stover, P.J. Safety of folic acid. Ann. N. Y. Acad. Sci. 2018, 1414, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, H.A. Losses of vitamins and trace minerals resulting from processing and preservation of foods. Am. J. Clin. Nutr. 1971, 24, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.A. Metformin and Vitamin B12 Deficiency: Where Do We Stand? J. Pharm. Pharm. Sci. 2016, 19, 382. [Google Scholar] [CrossRef] [Green Version]
- Shipton, M.J.; Thachil, J. Vitamin B 12 deficiency—A 21st century perspective. Clin. Med. 2015, 15, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and Recommendations about Total Homocysteine Determinations: An Expert Opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.H.; den Heijer, M.; Kluijtmans, L.A.J.; van den Heuve, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Van der Put, N.M.J.; Gabreëls, F.; Stevens, E.M.B.; Smeitink, J.A.M.; Trijbels, F.J.M.; Eskes, T.K.A.B.; van den Heuvel, L.P.; Blom, H.J. A Second Common Mutation in the Methylenetetrahydrofolate Reductase Gene: An Additional Risk Factor for Neural-Tube Defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- Hanson, N.Q.; Aras, O.; Yang, F.; Tsai, M.Y. C677T and A1298C polymorphisms of the methylenetetrahydrofolate reductase gene: Incidence and effect of combined genotypes on plasma fasting and post-methionine load homocysteine in vascular disease. Clin. Chem. 2001, 47, 661–666. [Google Scholar] [CrossRef] [Green Version]
- Boddie, A.M.; Steen, M.T.; Sullivan, K.M.; Pasquali, M.; Dembure, P.P.; Coates, R.J.; Elsas, L.J. Cystathionine-β-synthase deficiency: Detection of heterozygotes by the ratios of homocystein to cysteine and folate. Metabolism 1998, 47, 207–211. [Google Scholar] [CrossRef]
- Kenyon, S.H.; Nicolaou, A.; Gibbons, W.A. The Effect of Ethanol and Its Metabolites Upon Methionine Synthase Activity In Vitro. Alcohol 1998, 15, 305–309. [Google Scholar] [CrossRef]
- Jacques, P.F.; Bostom, A.G.; Wilson, P.W.F.; Rich, S.; Rosenberg, I.H.; Selhub, J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am. J. Clin. Nutr. 2001, 73, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, F.; Morisco, F.; Verde, V.; Ritieni, A.; Alezio, A.; Caporaso, N.; Fogliano, V. Moderate coffee consumption increases plasma glutathione but not homocysteine in healthy subjects. Aliment. Pharmacol. Ther. 2003, 17, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Danishpajooh, I.O.; Gudi, T.; Chen, Y.; Kharitonov, V.G.; Sharma, V.S.; Boss, G.R. Nitric Oxide Inhibits Methionine Synthase Activity in Vivo and Disrupts Carbon Flow through the Folate Pathway. J. Biol. Chem. 2001, 276, 27296–27303. [Google Scholar] [CrossRef] [Green Version]
- Subedi, H.; Brasch, N.E. Mechanistic Studies on the Reaction of Nitroxylcobalamin with Dioxygen: Evidence for Formation of a Peroxynitritocob(III)alamin Intermediate. Inorg. Chem. 2013, 52, 11608–11617. [Google Scholar] [CrossRef]
- Hultberg, B. Extracellular concentration of homocysteine in human cell lines is influenced by specific inhibitors of cyst(e)ine transport. Clin. Chem. Lab. Med. 2004, 42, 1–13. [Google Scholar] [CrossRef]
- Hultberg, B.; Andersson, A.; Isaksson, A. Higher export rate of homocysteine in a human endothelial cell line than in other human cell lines. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1448, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Refsum, H.; Guttormsen, A.B.; Fiskerstrand, T.; Ueland, P.M. On the Formation and Fate of Total Plasma Homocysteine. In Homocysteine Metabolism: From Basic Science to Clinical Medicine. Developments in Cardiovascular Medicine; Graham, I., Refsum, H., Rosenberg, I.H., Ueland, P.M., Shuman, J.M., Eds.; Springer: Boston, MA, USA, 1997; Volume 196, pp. 23–29. [Google Scholar]
- Selhub, J. Vitamin Status and Intake as Primary Determinants of Homocysteinemia in an Elderly Population. JAMA J. Am. Med. Assoc. 1993, 270, 2693. [Google Scholar] [CrossRef]
- Naurath, H.J.; Joosten, E.; Riezler, R.; Stabler, S.; Allen, R.H.; Lindenbaum, J. Effects of vitamin B12, folate, and vitamin B6 supplements in elderly people with normal serum vitamin concentrations. Lancet 1995, 346, 85–89. [Google Scholar] [CrossRef]
- Mansoor, M.A.; Ueland, P.M.; Aarsland, A.; Svardal, A.M. Redox status and protein binding of plasma homocysteine and other aminothiols in patients with homocystinuria. Metabolism 1993, 42, 1481–1485. [Google Scholar] [CrossRef]
- Guttormsen, A.B.; Schneede, J.; Ueland, P.M.; Refsum, H. Kinetics of total plasma homocysteine in subjects with hyperhomocysteinemia due to folate or cobalamin deficiency. Am. J. Clin. Nutr. 1996, 63, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Mudd, S.; Levy, H.; Skovby, F. Disorders of Transsulfuration. In The Metabolic and Molecular Bases of Inherited Disease, 7th ed.; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 1279–1327. [Google Scholar]
- Van Guldener, C.; Kulik, W.; Berger, R.; Dijkstra, D.A.; Jakobs, C.; Reijngoud, D.-J.; Donker, A.J.M.; Stehouwer, C.D.A.; De Meer, K. Homocysteine and methionine metabolism in ESRD: A stable isotope study. Kidney Int. 1999, 56, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Stam, F.; van Guldener, C.; ter Wee, P.M.; Kulik, W.; Smith, D.E.C.; Jakobs, C.; Stehouwer, C.D.A.; de Meer, K. Homocysteine clearance and methylation flux rates in health and end-stage renal disease: Association with S -adenosylhomocysteine. Am. J. Physiol. Physiol. 2004, 287, F215–F223. [Google Scholar] [CrossRef] [Green Version]
- Garibotto, G.; Valli, A.; Anderstam, B.; Eriksson, M.; Suliman, M.E.; Balbi, M.; Rollando, D.; Vigo, E.; Lindholm, B. The kidney is the major site of S-adenosylhomocysteine disposal in humans. Kidney Int. 2009, 76, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Kielstein, J.T.; Salpeter, S.R.; Buckley, N.S.; Cooke, J.P.; Fliser, D. Two Cardiovascular Risk Factors in One? Homocysteine and Its Relation to Glomerular Filtration Rate. Kidney Blood Press. Res. 2008, 31, 259–267. [Google Scholar] [CrossRef]
- Stam, F.; van Guldener, C.; Schalkwijk, C.G.; ter Wee, P.M.; Donker, A.J.M.; Stehouwer, C.D.A. Impaired renal function is associated with markers of endothelial dysfunction and increased inflammatory activity. Nephrol. Dial. Transplant. 2003, 18, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Bostom, A.G.; Shemin, D.; Lapane, K.L.; Miller, J.W.; Sutherland, P.; Nadeau, M.; Seyoum, E.; Hartman, W.; Prior, R.; Wilson, P.W.F.; et al. Hyperhomocysteinemia and traditional cardiovascular disease risk factors in end-stage renal disease patients on dialysis: A case-control study. Atherosclerosis 1995, 114, 93–103. [Google Scholar] [CrossRef]
- Shankar, A.; Wang, J.J.; Chua, B.; Rochtchina, E.; Flood, V.; Mitchell, P. Positive Association between Plasma Homocysteine Level and Chronic Kidney Disease. Kidney Blood Press. Res. 2008, 31, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Cahill, C.M.; Huang, X.; Roffman, J.L.; Lamon-Fava, S.; Fava, M.; Mischoulon, D.; Rogers, J.T. S-Adenosyl Methionine and Transmethylation Pathways in Neuropsychiatric Diseases Throughout Life. Neurotherapeutics 2018, 15, 156–175. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.; Banfield, K. S-Adenosylmethionine-Dependent Methyltransferases. In Homocysteine in Health and Disease; Carmel, R., Jacobsen, D., Eds.; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- Pfalzer, A.C.; Choi, S.-W.; Tammen, S.A.; Park, L.K.; Bottiglieri, T.; Parnell, L.D.; Lamon-Fava, S. S-adenosylmethionine mediates inhibition of inflammatory response and changes in DNA methylation in human macrophages. Physiol. Genom. 2014, 46, 617–623. [Google Scholar] [CrossRef]
- Jakubowski, H.; Głowacki, R. Chemical Biology of Homocysteine Thiolactone and Related Metabolites. Adv. Clin. Chem. 2011, 55, 81–103. [Google Scholar] [CrossRef]
- Förstermann, U. Janus-faced role of endothelial NO synthase in vascular disease: Uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol. Chem. 2006, 387, 1521–1533. [Google Scholar] [CrossRef]
- Moens, A.L.; Claeys, M.J.; Wuyts, F.L.; Goovaerts, I.; Van Hertbruggen, E.; Wendelen, L.C.; Van Hoof, V.O.; Vrints, C.J. Effect of Folic Acid on Endothelial Function Following Acute Myocardial Infarction. Am. J. Cardiol. 2007, 99, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Sass, J.O.; Nakanishi, T.; Sato, T.; Sperl, W.; Shimizu, A. S-Homocysteinylation of transthyretin is detected in plasma and serum of humans with different types of hyperhomocysteinemia. Biochem. Biophys. Res. Commun. 2003, 310, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullan, L.M.; Olney, J.W.; Price, M.T.; Compton, R.P.; Hood, W.F.; Michel, J.; Monahan, J.B. Excitatory Amino Acid Receptor Potency and Subclass Specificity of Sulfur-Containing Amino Acids. J. Neurochem. 1987, 49, 1301–1307. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Protein and Amino Acid Requirements in Human Nutrition; WHO Technical Report Series No. 935; Report of a Joint WHO/FAO/UNU Expert Consultation; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Krupková-Meixnerová, L.; Veselá, K.; Vitová, A.; Janosíková, B.; Andel, M.; Kozich, V. Methionine-loading test: Evaluation of adverse effects and safety in an epidemiological study. Clin. Nutr. 2002, 21, 151–156. [Google Scholar] [CrossRef]
- Bostom, A.G.; Jacques, P.F.; Nadeau, M.R.; Williams, R.R.; Ellison, R.C.; Selhub, J. Post-methionine load hyperhomocysteinemia in persons with normal fasting total plasma homocysteine: Initial results from The NHLBI Family Heart Study. Atherosclerosis 1995, 116, 147–151. [Google Scholar] [CrossRef]
- Kies, C.; Fox, H.; Aprahamian, S. Comparative Value of L-, DL-, and D-Methionine Supplementation of an Oat-based Diet for Humans. J. Nutr. 1975, 105, 809–814. [Google Scholar] [CrossRef]
- Harter, J.M.; Baker, D.H. Sulfur Amino Acid Activity of d- and l-Homocysteine for Chicks. Exp. Biol. Med. 1978, 157, 139–143. [Google Scholar] [CrossRef]
- Baker, D.H. Utilization of Precursors for l-Amino Acids. In Amino Acids in Farm Animal Nutrition; D’Mello, J.P.F., Ed.; CAB International: Wallingford, UK, 1994; pp. 37–61. [Google Scholar]
- Kožich, V.; Ditrói, T.; Sokolová, J.; Křížková, M.; Krijt, J.; Ješina, P.; Nagy, P. Metabolism of sulfur compounds in homocystinurias. Br. J. Pharmacol. 2019, 176, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I.P.; Lee, P.J.; Briddon, A. Homocysteine and cysteine-albumin binding in homocystinuria: Assessment of cysteine status and implications for glutathione synthesis? Amino Acids 2002, 22, 109–118. [Google Scholar] [CrossRef]
- Pan, L.L.; Liu, X.H.; Gong, Q.H.; Yang, H.B.; Zhu, Y.Z. Role of Cystathionine γ-Lyase/Hydrogen Sulfide Pathway in Cardiovascular Disease: A Novel Therapeutic Strategy? Antioxid. Redox Signal. 2012, 17, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.S.; Mishra, P.K. H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 2017, 7, 3639. [Google Scholar] [CrossRef]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [Green Version]
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyperhomocysteinemia as a Risk Factor for Vascular Contributions to Cognitive Impairment and Dementia. Front. Aging Neurosci. 2018, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its Possible Emerging Role in At-Risk Population Groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [Green Version]
- Jernerén, F.; Elshorbagy, A.K.; Oulhaj, A.; Smith, S.M.; Refsum, H.; Smith, A.D. Brain atrophy in cognitively impaired elderly: The importance of long-chain ω-3 fatty acids and B vitamin status in a randomized controlled trial. Am. J. Clin. Nutr. 2015, 102, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Soininen, H.; Solomon, A.; Visser, P.J.; Hendrix, S.B.; Blennow, K.; Kivipelto, M.; Hartmann, T.; Hallikainen, I.; Hallikainen, M.; Helisalmi, S.; et al. 24-month intervention with a specific multinutrient in people with prodromal Alzheimer’s disease (LipiDiDiet): A randomised, double-blind, controlled trial. Lancet Neurol. 2017, 16, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, R.A.; Fuso, A.; Nicolia, V.; Scarpa, S. S-Adenosylmethionine Prevents Oxidative Stress and Modulates Glutathione Metabolism in TgCRND8 Mice Fed a B-Vitamin Deficient Diet. J. Alzheimer’s Dis. 2010, 20, 997–1002. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-β deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef]
- Nieraad, H.; de Bruin, N.; Arne, O.; Hofmann, M.C.J.; Schmidt, M.; Saito, T.; Saido, T.C.; Gurke, R.; Schmidt, D.; Till, U.; et al. Impact of Hyperhomocysteinemia and Different Dietary Interventions on Cognitive Performance in a Knock-in Mouse Model for Alzheimer’s Disease. Nutrients 2020, 12, 3248. [Google Scholar] [CrossRef]
- Javelot, H.; Messaoudi, M.; Jacquelin, C.; Bisson, J.F.; Rozan, P.; Nejdi, A.; Lazarus, C.; Cassel, J.C.; Strazielle, C.; Lalonde, R. Behavioral and neurochemical effects of dietary methyl donor deficiency combined with unpredictable chronic mild stress in rats. Behav. Brain Res. 2014, 261, 8–16. [Google Scholar] [CrossRef]
- Schwahn, B.C.; Wendel, U.; Lussier-Cacan, S.; Mar, M.-H.; Zeisel, S.H.; Leclerc, D.; Castro, C.; Garrow, T.A.; Rozen, R. Effects of betaine in a murine model of mild cystathionine-β-synthase deficiency. Metabolism 2004, 53, 594–599. [Google Scholar] [CrossRef]
- Bhatia, P.; Singh, N. Ameliorative effect of ozagrel, a thromboxane A2 synthase inhibitor, in hyperhomocysteinemia-induced experimental vascular cognitive impairment and dementia. Fundam. Clin. Pharmacol. 2020, 35, 650–666. [Google Scholar] [CrossRef]
- Troen, A.M.; Shukitt-Hale, B.; Chao, W.; Albuquerque, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. The cognitive impact of nutritional homocysteinemia in Apolipoprotein-E deficient mice. J. Alzheimer’s Dis. 2006, 9, 381–392. [Google Scholar] [CrossRef]
- Selhub, J.; Troen, A.M. Sulfur amino acids and atherosclerosis: A role for excess dietary methionine. Ann. N. Y. Acad. Sci. 2016, 1363, 18–25. [Google Scholar] [CrossRef]
- Devlin, A.M.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Rozen, R.; Lentz, S.R. Effect of Mthfr genotype on diet-induced hyperhomocysteinemia and vascular function in mice. Blood 2004, 103, 2624–2629. [Google Scholar] [CrossRef] [Green Version]
- Pirchl, M.; Ullrich, C.; Humpel, C. Differential effects of short- and long-term hyperhomocysteinaemia on cholinergic neurons, spatial memory and microbleedings in vivo in rats. Eur. J. Neurosci. 2010, 32, 1516–1527. [Google Scholar] [CrossRef]
- Stead, L.M.; Au, K.P.; Jacobs, R.L.; Brosnan, M.E.; Brosnan, J.T. Methylation demand and homocysteine metabolism: Effects of dietary provision of creatine and guanidinoacetate. Am. J. Physiol. Metab. 2001, 281, E1095–E1100. [Google Scholar] [CrossRef]
- Basu, T.K.; Mann, S. Vitamin B-6 Normalizes the Altered Sulfur Amino Acid Status of Rats Fed Diets Containing Pharmacological Levels of Niacin without Reducing Niacin’s Hypolipidemic Effects. J. Nutr. 1997, 127, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalska, M.; Tothova, B.; Kovalska, L.; Tatarkova, Z.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Association of Induced Hyperhomocysteinemia with Alzheimer’s Disease-Like Neurodegeneration in Rat Cortical Neurons After Global Ischemia-Reperfusion Injury. Neurochem. Res. 2018, 43, 1766–1778. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.A.; Riedel, G.; Algaidi, S.A.; Whalley, L.J.; Platt, B. Enhanced hippocampal long-term potentiation in rats after chronic exposure to homocysteine. Neurosci. Lett. 2005, 373, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Rabaneda, L.G.; Carrasco, M.; López-Toledano, M.A.; Murillo-Carretero, M.; Ruiz, F.A.; Estrada, C.; Castro, C. Homocysteine inhibits proliferation of neuronal precursors in the mouse adult brain by impairing the basic fibroblast growth factor signaling cascade and reducing extracellular regulated kinase 1/2-dependent cyclin E expression. FASEB J. 2008, 22, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Elsherbiny, N.M.; Sharma, I.; Kira, D.; Alhusban, S.; Samra, Y.A.; Jadeja, R.; Martin, P.; Al-Shabrawey, M.; Tawfik, A. Homocysteine Induces Inflammation in Retina and Brain. Biomolecules 2020, 10, 393. [Google Scholar] [CrossRef] [Green Version]
- Cole, P.D.; Vijayanathan, V.; Ali, N.F.; Wagshul, M.E.; Tanenbaum, E.J.; Price, J.; Dalal, V.; Gulinello, M.E. Memantine Protects Rats Treated with Intrathecal Methotrexate from Developing Spatial Memory Deficits. Clin. Cancer Res. 2013, 19, 4446–4454. [Google Scholar] [CrossRef] [Green Version]
- Fukada, S.; Shimada, Y.; Morita, T.; Sugiyama, K. Suppression of Methionine-Induced Hyperhomocysteinemia by Glycine and Serine in Rats. Biosci. Biotechnol. Biochem. 2006, 70, 2403–2409. [Google Scholar] [CrossRef] [Green Version]
- Akahoshi, N.; Kobayashi, C.; Ishizaki, Y.; Izumi, T.; Himi, T.; Suematsu, M.; Ishii, I. Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine β-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum. Mol. Genet. 2008, 17, 1994–2005. [Google Scholar] [CrossRef] [Green Version]
- Rhodehouse, B.C.; Erickson, M.A.; Banks, W.A.; Bearden, S.E. Hyperhomocysteinemic Mice Show Cognitive Impairment Without Features of Alzheimer’s Disease Phenotype. J. Alzheimer’s Dis. 2013, 35, 59–66. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Deng, L.; Leclerc, D.; Malysheva, O.; Bedell, B.J.; Caudill, M.A.; Rozen, R. Severe methylenetetrahydrofolate reductase deficiency in mice results in behavioral anomalies with morphological and biochemical changes in hippocampus. Mol. Genet. Metab. 2012, 106, 149–159. [Google Scholar] [CrossRef]
- Sørensen, J.T.; Gaustadnes, M.; Stabler, S.P.; Allen, R.H.; Mudd, S.H.; Hvas, A.-M. Molecular and biochemical investigations of patients with intermediate or severe hyperhomocysteinemia. Mol. Genet. Metab. 2016, 117, 344–350. [Google Scholar] [CrossRef]
- Román, G.; Mancera-Páez, O.; Bernal, C. Epigenetic Factors in Late-Onset Alzheimer’s Disease: MTHFR and CTH Gene Polymorphisms, Metabolic Transsulfuration and Methylation Pathways, and B Vitamins. Int. J. Mol. Sci. 2019, 20, 319. [Google Scholar] [CrossRef] [Green Version]
- Akahoshi, N.; Yokoyama, A.; Nagata, T.; Miura, A.; Kamata, S.; Ishii, I. Abnormal Amino Acid Profiles of Blood and Cerebrospinal Fluid from Cystathionine β-Synthase-Deficient Mice, an Animal Model of Homocystinuria. Biol. Pharm. Bull. 2019, 42, 1054–1057. [Google Scholar] [CrossRef]
- Prieur, E.A.K.; Pjetri, E.; Zeisel, S.H.; Jadavji, N.M. Reduced brain volume and impaired memory in betaine homocysteine S -methyltransferase knockout mice. Appl. Physiol. Nutr. Metab. 2017, 42, 1228–1231. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Bahous, R.H.; Deng, L.; Malysheva, O.; Grand’maison, M.; Bedell, B.J.; Caudill, M.A.; Rozen, R. Mouse model for deficiency of methionine synthase reductase exhibits short-term memory impairment and disturbances in brain choline metabolism. Biochem. J. 2014, 461, 205–212. [Google Scholar] [CrossRef]
- Yakovleva, O.; Bogatova, K.; Mukhtarova, R.; Yakovlev, A.; Shakhmatova, V.; Gerasimova, E.; Ziyatdinova, G.; Hermann, A.; Sitdikova, G. Hydrogen Sulfide Alleviates Anxiety, Motor, and Cognitive Dysfunctions in Rats with Maternal Hyperhomocysteinemia via Mitigation of Oxidative Stress. Biomolecules 2020, 10, 995. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, Y.; Sun, J.; Zhang, Y.; Luo, T.; Li, B.; Jiang, Y.; Shi, Y.; Le, G. Dietary methionine restriction ameliorates the impairment of learning and memory function induced by obesity in mice. Food Funct. 2019, 10, 1411–1425. [Google Scholar] [CrossRef]
- Xie, F.; Zhao, Y.; Ma, J.; Gong, J.-B.; Wang, S.-D.; Zhang, L.; Gao, X.-J.; Qian, L.-J. The involvement of homocysteine in stress-induced Aβ precursor protein misprocessing and related cognitive decline in rats. Cell Stress Chaperones 2016, 21, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Kamat, P.K.; Mallonee, C.J.; George, A.K.; Tyagi, S.C.; Tyagi, N. Homocysteine, Alcoholism, and Its Potential Epigenetic Mechanism. Alcohol. Clin. Exp. Res. 2016, 40, 2474–2481. [Google Scholar] [CrossRef]
- Sinha, M.; Saha, A.; Basu, S.; Pal, K.; Chakrabarti, S. Aging and antioxidants modulate rat brain levels of homocysteine and dehydroepiandrosterone sulphate (DHEA-S): Implications in the pathogenesis of Alzheimer’s disease. Neurosci. Lett. 2010, 483, 123–126. [Google Scholar] [CrossRef]
- Singh, S.K.; Misra, U.K.; Kalita, J.; Bora, H.K.; Murthy, R.C. Nitrous oxide related behavioral and histopathological changes may be related to oxidative stress. Neurotoxicology 2015, 48, 44–49. [Google Scholar] [CrossRef]
- Borai, I.H.; Ezz, M.K.; Rizk, M.Z.; Aly, H.F.; El-Sherbiny, M.; Matloub, A.A.; Fouad, G.I. Therapeutic impact of grape leaves polyphenols on certain biochemical and neurological markers in AlCl3-induced Alzheimer’s disease. Biomed. Pharmacother. 2017, 93, 837–851. [Google Scholar] [CrossRef]
- Ismail, M.F.; ElMeshad, A.; Salem, N. Potential therapeutic effect of nanobased formulation of rivastigmine on rat model of Alzheimer’s disease. Int. J. Nanomed. 2013, 8, 393. [Google Scholar] [CrossRef]
- El-Missiry, M.A.; Othman, A.I.; El-Sawy, M.R.; Lebede, M.F. Neuroprotective effect of epigallocatechin-3-gallate (EGCG) on radiation-induced damage and apoptosis in the rat hippocampus. Int. J. Radiat. Biol. 2018, 94, 798–808. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S. Fatty acids rehabilitated long-term neurodegenerative: Like symptoms in olfactory bulbectomized rats. J. Neural Transm. 2015, 122, 629–641. [Google Scholar] [CrossRef]
- Singh, M.; Prakash, A. Possible role of endothelin receptor against hyperhomocysteinemia and β-amyloid induced AD type of vascular dementia in rats. Brain Res. Bull. 2017, 133, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Nieraad, H.; de Bruin, N.; Arne, O.; Hofmann, M.C.J.; Gurke, R.; Schmidt, D.; Ritter, M.; Parnham, M.J.; Geisslinger, G. Effects of Alzheimer-Like Pathology on Homocysteine and Homocysteic Acid Levels—An Exploratory In Vivo Kinetic Study. Int. J. Mol. Sci. 2021, 22, 927. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Maxwell, R.R.; Wolf, A.J.; Spira, M.; Gulinello, M.E.; Cole, P.D. Methotrexate causes persistent deficits in memory and executive function in a juvenile animal model. Neuropharmacology 2018, 139, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, S.O.; Heimfarth, L.; Pelaez, P.d.L.; Vanzin, C.S.; Viana, L.; Wyse, A.T.S.; Pessoa-Pureur, R. Homocysteine activates calcium-mediated cell signaling mechanisms targeting the cytoskeleton in rat hippocampus. Int. J. Dev. Neurosci. 2008, 26, 447–455. [Google Scholar] [CrossRef]
- Lockhart, B.; Jones, C.; Cuisinier, C.; Villain, N.; Peyroulan, D.; Lestage, P. Inhibition of l-homocysteic acid and buthionine sulphoximine-mediated neurotoxicity in rat embryonic neuronal cultures with α-lipoic acid enantiomers. Brain Res. 2000, 855, 292–297. [Google Scholar] [CrossRef]
- Takeuchi, H.; Iba, M.; Inoue, H.; Higuchi, M.; Takao, K.; Tsukita, K.; Karatsu, Y.; Iwamoto, Y.; Miyakawa, T.; Suhara, T.; et al. P301S Mutant Human Tau Transgenic Mice Manifest Early Symptoms of Human Tauopathies with Dementia and Altered Sensorimotor Gating. PLoS ONE 2011, 6, e21050. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, J.-Z.; Xu, X.-Y.; Hu, J.; Xu, T.; Jin, S.; Yang, S.-J.; Wang, J.-Z. Liraglutide Ameliorates Hyperhomocysteinemia-Induced Alzheimer-Like Pathology and Memory Deficits in Rats via Multi-molecular Targeting. Neurosci. Bull. 2019, 35, 724–734. [Google Scholar] [CrossRef]
- Zeng, P.; Shi, Y.; Wang, X.-M.; Lin, L.; Du, Y.-J.; Tang, N.; Wang, Q.; Fang, Y.-Y.; Wang, J.-Z.; Zhou, X.-W.; et al. Emodin Rescued Hyperhomocysteinemia-Induced Dementia and Alzheimer’s Disease-Like Features in Rats. Int. J. Neuropsychopharmacol. 2019, 22, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Mullane, K.; Williams, M. Preclinical Models of Alzheimer’s Disease: Relevance and Translational Validity. Curr. Protoc. Pharmacol. 2019, 84, e57. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Ilango, K.; Singh, P.K.; Karmakar, D.; Singh, G.P.I.; Kumari, R.; Dubey, G.P. Age dependent levels of plasma homocysteine and cognitive performance. Behav. Brain Res. 2015, 283, 139–144. [Google Scholar] [CrossRef]
- Görtz, P.; Hoinkes, A.; Fleischer, W.; Otto, F.; Schwahn, B.; Wendel, U.; Siebler, M. Implications for hyperhomocysteinemia: Not homocysteine but its oxidized forms strongly inhibit neuronal network activity. J. Neurol. Sci. 2004, 218, 109–114. [Google Scholar] [CrossRef]
- Sommer, S.; Hunzinger, C.; Schillo, S.; Klemm, M.; Biefang-Arndt, K.; Schwall, G.; Pütter, S.; Hoelzer, K.; Schroer, K.; Stegmann, W.; et al. Molecular Analysis of Homocysteic Acid-Induced Neuronal Stress. J. Proteome Res. 2004, 3, 572–581. [Google Scholar] [CrossRef]
- Vladychenskaya, E.A.; Tyulina, O.V.; Boldyrev, A.A. Effect of homocysteine and homocysteic acid on glutamate receptors on rat lymphocytes. Bull. Exp. Biol. Med. 2006, 142, 47–50. [Google Scholar] [CrossRef]
- Hasegawa, T.; Kosoku, Y.; Sano, Y.; Yoshida, H.; Kudoh, C.; Tabira, T. Homocysteic Acid in Blood Can Detect Mild Cognitive Impairment: A Preliminary Study. J. Alzheimer’s Dis. 2020, 77, 773–780. [Google Scholar] [CrossRef]
- Hasegawa, T.; Mikoda, N.; Kitazawa, M.; LaFerla, F.M. Treatment of Alzheimer’s Disease with Anti-Homocysteic Acid Antibody in 3xTg-AD Male Mice. PLoS ONE 2010, 5, e8593. [Google Scholar] [CrossRef]
- Ford, A.H.; Almeida, O.P. Effect of Vitamin B Supplementation on Cognitive Function in the Elderly: A Systematic Review and Meta-Analysis. Drugs Aging 2019, 36, 419–434. [Google Scholar] [CrossRef]
- Mielech, A.; Puścion-Jakubik, A.; Markiewicz-Żukowska, R.; Socha, K. Vitamins in Alzheimer’s Disease—Review of the Latest Reports. Nutrients 2020, 12, 3458. [Google Scholar] [CrossRef]
- McCleery, J.; Abraham, R.P.; Denton, D.A.; Rutjes, A.W.S.; Chong, L.-Y.; Al-Assaf, A.S.; Griffith, D.J.; Rafeeq, S.; Yaman, H.; Malik, M.A.; et al. Vitamin and mineral supplementation for preventing dementia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst. Rev. 2018, 2018. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma Homocysteine as a Risk Factor for Dementia and Alzheimer’s Disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef]
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and Dementia: An International Consensus Statement1. J. Alzheimer’s Dis. 2018, 62, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Kim, S.Y.; Sok, S.R. Effects of Multivitamin Supplements on Cognitive Function, Serum Homocysteine Level, and Depression of Korean Older Adults with Mild Cognitive Impairment in Care Facilities. J. Nurs. Scholarsh. 2016, 48, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-Lowering by B Vitamins Slows the Rate of Accelerated Brain Atrophy in Mild Cognitive Impairment: A Randomized Controlled Trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, E.; Refsum, H.; Tell, G.S.; Engedal, K.; Vollset, S.E.; Ueland, P.M.; Nygaard, H.A.; Smith, A.D. Plasma total homocysteine and memory in the elderly: The Hordaland Homocysteine study. Ann. Neurol. 2005, 58, 847–857. [Google Scholar] [CrossRef]
- Jager, C.A.; Oulhaj, A.; Jacoby, R.; Refsum, H.; Smith, A.D. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2012, 27, 592–600. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic studies of modifiable factors associated with cognition and dementia: Systematic review and meta-analysis. BMC Public Health 2014, 14, 643. [Google Scholar] [CrossRef] [Green Version]
- Farina, N.; Jernerén, F.; Turner, C.; Hart, K.; Tabet, N. Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Exp. Gerontol. 2017, 99, 146–150. [Google Scholar] [CrossRef]
- Bonetti, F.; Brombo, G.; Magon, S.; Zuliani, G. Cognitive Status According to Homocysteine and B-Group Vitamins in Elderly Adults. J. Am. Geriatr. Soc. 2015, 63, 1158–1163. [Google Scholar] [CrossRef]
- Cheng, D.; Kong, H.; Pang, W.; Yang, H.; Lu, H.; Huang, C.; Jiang, Y. B vitamin supplementation improves cognitive function in the middle aged and elderly with hyperhomocysteinemia. Nutr. Neurosci. 2016, 19, 461–466. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.-X.; Miller, J.; Green, R.; Luchsinger, J.A. Plasma Homocysteine and Risk of Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. 2009, 27, 11–17. [Google Scholar] [CrossRef]
- Behrens, A.; Graessel, E.; Pendergrass, A.; Donath, C. Vitamin B—Can it prevent cognitive decline? A systematic review and meta-analysis. Syst. Rev. 2020, 9, 111. [Google Scholar] [CrossRef]
- Kwok, T.; Wu, Y.; Lee, J.; Lee, R.; Yung, C.Y.; Choi, G.; Lee, V.; Harrison, J.; Lam, L.; Mok, V. A randomized placebo-controlled trial of using B vitamins to prevent cognitive decline in older mild cognitive impairment patients. Clin. Nutr. 2020, 39, 2399–2405. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.; Macpherson, H.; Pipingas, A. Improved Blood Biomarkers but No Cognitive Effects from 16 Weeks of Multivitamin Supplementation in Healthy Older Adults. Nutrients 2015, 7, 3796–3812. [Google Scholar] [CrossRef] [Green Version]
- Tabet, N.; Rafi, H.; Weaving, G.; Lyons, B.; Iversen, S.A. Behavioural and psychological symptoms of Alzheimer type dementia are not correlated with plasma homocysteine concentration. Dement. Geriatr. Cogn. Disord. 2006, 22, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-M.; Ye, J.-X.; Mu, J.-S.; Cui, X.-P. Efficacy of Vitamin B Supplementation on Cognition in Elderly Patients with Cognitive-Related Diseases. J. Geriatr. Psychiatry Neurol. 2017, 30, 50–59. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.A.; Green, T.J.; Skeaff, C.M.; Knight, R.G.; Mann, J.I.; Williams, S.M. A Controlled Trial of Homocysteine Lowering and Cognitive Performance. N. Engl. J. Med. 2006, 354, 2764–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.D.; Refsum, H. Homocysteine, B Vitamins, and Cognitive Impairment. Ann. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Mlinarić, A.; Horvat, M.; Šupak Smolčić, V. Dealing with the positive publication bias: Why you should really publish your negative results. Biochem. Med. 2017, 27, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, E.S.; van der Worp, H.B.; Bath, P.M.W.; Howells, D.W.; Macleod, M.R. Publication Bias in Reports of Animal Stroke Studies Leads to Major Overstatement of Efficacy. PLoS Biol. 2010, 8, e1000344. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Parekh, S.; Hooper, L.; Loke, Y.; Ryder, J.; Sutton, A.; Hing, C.; Kwok, C.; Pang, C.; Harvey, I. Dissemination and publication of research findings: An updated review of related biases. Health Technol. Assess. 2010, 14, 1–220. [Google Scholar] [CrossRef]
- Macleod, M.R.; Lawson McLean, A.; Kyriakopoulou, A.; Serghiou, S.; de Wilde, A.; Sherratt, N.; Hirst, T.; Hemblade, R.; Bahor, Z.; Nunes-Fonseca, C.; et al. Risk of Bias in Reports of In Vivo Research: A Focus for Improvement. PLoS Biol. 2015, 13, e1002273. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Avey, M.T.; Moher, D.; Sullivan, K.J.; Fergusson, D.; Griffin, G.; Grimshaw, J.M.; Hutton, B.; Lalu, M.M.; Macleod, M.; Marshall, J.; et al. The Devil Is in the Details: Incomplete Reporting in Preclinical Animal Research. PLoS ONE 2016, 11, e0166733. [Google Scholar] [CrossRef] [Green Version]
- Bespalov, A.; Steckler, T.; Skolnick, P. Be positive about negatives–recommendations for the publication of negative (or null) results. Eur. Neuropsychopharmacol. 2019, 29, 1312–1320. [Google Scholar] [CrossRef]
- Vollert, J.; Schenker, E.; Macleod, M.; Bespalov, A.; Wuerbel, H.; Michel, M.; Dirnagl, U.; Potschka, H.; Waldron, A.-M.; Wever, K.; et al. Systematic review of guidelines for internal validity in the design, conduct and analysis of preclinical biomedical experiments involving laboratory animals. BMJ Open Sci. 2020, 4, e100046. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [Green Version]
- Schächinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic Impact of Coronary Vasodilator Dysfunction on Adverse Long-Term Outcome of Coronary Heart Disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [Green Version]
- Wald, D.S. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. BMJ 2002, 325, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Homocysteine Studies Collaboration Homocysteine and Risk of Ischemic Heart Disease and Stroke. JAMA 2002, 288, 2015. [CrossRef]
- Bhargava, S.; Parakh, R.; Manocha, A.; Ali, A.; Srivastava, L.M. Prevalence of Hyperhomocysteinemia in Vascular Disease: Comparative Study of Thrombotic Venous Disease Vis-è-Vis Occlusive Arterial Disease. Vascular 2007, 15, 149–153. [Google Scholar] [CrossRef]
- Wanner, C.; Amann, K.; Shoji, T. The heart and vascular system in dialysis. Lancet 2016, 388, 276–284. [Google Scholar] [CrossRef]
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert Rev. Cardiovasc. Ther. 2018, 16, 559–565. [Google Scholar] [CrossRef]
- Spence, J. Nutrition and Risk of Stroke. Nutrients 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic Acid Therapy and Cardiovascular Disease in ESRD or Advanced Chronic Kidney Disease: A Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 482–488. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- De Jager, J.; Kooy, A.; Lehert, P.; Wulffele, M.G.; van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.M.; Stehouwer, C.D.A. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ 2010, 340, c2181. [Google Scholar] [CrossRef] [Green Version]
- Audelin, M.C.; Genest, J. Homocysteine and cardiovascular disease in diabetes mellitus. Atherosclerosis 2001, 159, 497–511. [Google Scholar] [CrossRef]
- Lind, M.V.; Lauritzen, L.; Kristensen, M.; Ross, A.B.; Eriksen, J.N. Effect of folate supplementation on insulin sensitivity and type 2 diabetes: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2019, 109, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Wile, D.J.; Toth, C. Association of Metformin, Elevated Homocysteine, and Methylmalonic Acid Levels and Clinically Worsened Diabetic Peripheral Neuropathy. Diabetes Care 2010, 33, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Heijer, M.; Lewington, S.; Clarke, R. Homocysteine, MTHFR and risk of venous thrombosis: A meta-analysis of published epidemiological studies. J. Thromb. Haemost. 2005, 3, 292–299. [Google Scholar] [CrossRef]
- Zhou, K.; Zhao, R.; Geng, Z.; Jiang, L.; Cao, Y.; Xu, D.; Liu, Y.; Huang, L.; Zhou, J. Association between B-group vitamins and venous thrombosis: Systematic review and meta-analysis of epidemiological studies. J. Thromb. Thrombolysis 2012, 34, 459–467. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Selhub, J.; Miletich, J.P.; Malinow, M.R.; Stampfer, M.J. Interrelation of Hyperhomocyst(e)inemia, Factor V Leiden, and Risk of Future Venous Thromboembolism. Circulation 1997, 95, 1777–1782. [Google Scholar] [CrossRef]
- Bjelland, I.; Tell, G.S.; Vollset, S.E.; Refsum, H.; Ueland, P.M. Folate, Vitamin B12, Homocysteine, and the MTHFR 677C→T Polymorphism in Anxiety and Depression. Arch. Gen. Psychiatry 2003, 60, 618. [Google Scholar] [CrossRef] [Green Version]
- Skarupski, K.A.; Tangney, C.; Li, H.; Ouyang, B.; Evans, D.A.; Morris, M.C. Longitudinal association of vitamin B-6, folate, and vitamin B-12 with depressive symptoms among older adults over time. Am. J. Clin. Nutr. 2010, 92, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Almeida, O.P.; Marsh, K.; Alfonso, H.; Flicker, L.; Davis, T.M.E.; Hankey, G.J. B-vitamins reduce the long-term risk of depression after stroke: The VITATOPS-DEP trial. Ann. Neurol. 2010, 68, 503–510. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E. Homocysteine and levodopa: Should Parkinson disease patients receive preventative therapy? Neurology 2004, 63, 886–891. [Google Scholar] [CrossRef]
- Fathe, K.; Palacios, A.; Finnell, R.H. Brief report novel mechanism for valproate-induced teratogenicity. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Stanger, O.; Fowler, B.; Piertzik, K.; Huemer, M.; Haschke-Becher, E.; Semmler, A.; Lorenzl, S.; Linnebank, M. Homocysteine, folate and vitamin B 12 in neuropsychiatric diseases: Review and treatment recommendations. Expert Rev. Neurother. 2009, 9, 1393–1412. [Google Scholar] [CrossRef] [Green Version]
- Miziak, B.; Chrościńska-Krawczyk, M.; Czuczwar, S.J. An update on the problem of osteoporosis in people with epilepsy taking antiepileptic drugs. Expert Opin. Drug Saf. 2019, 18, 679–689. [Google Scholar] [CrossRef]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and Neurologic Disorders: A Review. J. Clin. Neurol. 2014, 10, 281. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, E.H.; Green, R. Valproate and folate: Congenital and developmental risks. Epilepsy Behav. 2020, 108, 107068. [Google Scholar] [CrossRef]
- Li, J.; Parker, B.; Martyn, C.; Natarajan, C.; Guo, J. The PMP22 Gene and Its Related Diseases. Mol. Neurobiol. 2013, 47, 673–698. [Google Scholar] [CrossRef] [Green Version]
- Morkbak, A.L.; Hvas, A.-M.; Milman, N.; Nexo, E. Holotranscobalamin remains unchanged during pregnancy. Longitudinal changes of cobalamins and their binding proteins during pregnancy and postpartum. Haematologica 2007, 92, 1711–1712. [Google Scholar] [CrossRef]
- Vollset, S.E.; Refsum, H.; Irgens, L.M.; Emblem, B.M.; Tverdal, A.; Gjessing, H.K.; Monsen, A.L.B.; Ueland, P.M. Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: The Hordaland Homocysteine Study. Am. J. Clin. Nutr. 2000, 71, 962–968. [Google Scholar] [CrossRef]
- Kalhan, S.C. One carbon metabolism in pregnancy: Impact on maternal, fetal and neonatal health. Mol. Cell. Endocrinol. 2016, 435, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Gris, J.-C.; Perneger, T.V.; Quéré, I.; Mercier, E.; Fabbro-Peray, P.; Lavigne-Lissalde, G.; Hoffet, M.; Déchaud, H.; Boyer, J.-C.; Ripart-Neveu, S.; et al. Antiphospholipid/antiprotein antibodies, hemostasis-related autoantibodies, and plasma homocysteine as risk factors for a first early pregnancy loss: A matched case-control study. Blood 2003, 102, 3504–3513. [Google Scholar] [CrossRef] [Green Version]
- Quéré, I. Vitamin supplementation and pregnancy outcome in women with recurrent early pregnancy loss and hyperhomocysteinemia. Fertil. Steril. 2001, 75, 823–825. [Google Scholar] [CrossRef]
- Imbard, A.; Benoist, J.-F.; Blom, H. Neural Tube Defects, Folic Acid and Methylation. Int. J. Environ. Res. Public Health 2013, 10, 4352–4389. [Google Scholar] [CrossRef] [Green Version]
- Lamers, Y.; Prinz-Langenohl, R.; Brämswig, S.; Pietrzik, K. Red blood cell folate concentrations increase more after supplementation with [6 S]-5-methyltetrahydrofolate than with folic acid in women of childbearing age. Am. J. Clin. Nutr. 2006, 84, 156–161. [Google Scholar] [CrossRef]
- Lumley, J.; Watson, L.; Watson, M.; Bower, C. Periconceptional Supplementation with Folate and/or Multivitamins for Preventing Neural Tube Defects. In Cochrane Database of Systematic Reviews; Lumley, J., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar]
- Czeizel, A.E. The primary prevention of birth defects: Multivitamins or folic acid? Int. J. Med. Sci. 2004, 1, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Molloy, A.M. Genetic Aspects of Folate Metabolism; Springer: Berlin/Heidelberg, Germany, 2012; pp. 105–130. [Google Scholar]
- Rogne, T.; Tielemans, M.J.; Chong, M.F.-F.; Yajnik, C.S.; Krishnaveni, G.V.; Poston, L.; Jaddoe, V.W.V.; Steegers, E.A.P.; Joshi, S.; Chong, Y.-S.; et al. Associations of Maternal Vitamin B12 Concentration in Pregnancy with the Risks of Preterm Birth and Low Birth Weight: A Systematic Review and Meta-Analysis of Individual Participant Data. Am. J. Epidemiol. 2017, 185, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Venkatramanan, S.; Armata, I.E.; Strupp, B.J.; Finkelstein, J.L. Vitamin B-12 and Cognition in Children. Adv. Nutr. 2016, 7, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Adaikalakoteswari, A.; Wood, C.; Mina, T.H.; Webster, C.; Goljan, I.; Weldeselassie, Y.; Reynolds, R.M.; Saravanan, P. Vitamin B12 deficiency and altered one-carbon metabolites in early pregnancy is associated with maternal obesity and dyslipidaemia. Sci. Rep. 2020, 10, 11066. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Kumar, K.A.; Basak, T.; Bhardwaj, G.; Yadav, D.K.; Lalitha, A.; Chandak, G.R.; Raghunath, M.; Sengupta, S. PPAR signaling pathway is a key modulator of liver proteome in pups born to vitamin B12 deficient rats. J. Proteom. 2013, 91, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.A.; Nédélec, E.; Schroeder, H.; Alberto, J.-M.; Bossenmeyer-Pourié, C.; Guéant, J.-L.; Daval, J.-L. Gestational Vitamin B Deficiency Leads to Homocysteine-Associated Brain Apoptosis and Alters Neurobehavioral Development in Rats. Am. J. Pathol. 2007, 170, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezo, Y.J.R.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Tawfik, A.; Mohamed, R.; Elsherbiny, N.; DeAngelis, M.; Bartoli, M.; Al-Shabrawey, M. Homocysteine: A Potential Biomarker for Diabetic Retinopathy. J. Clin. Med. 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Lee, H.; Kim, J.-M.; Chae, E.H.; Kim, S.J.; Chang, N. Short-Term Hyperhomocysteinemia-Induced Oxidative Stress Activates Retinal Glial Cells and Increases Vascular Endothelial Growth Factor Expression in Rat Retina. Biosci. Biotechnol. Biochem. 2007, 71, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Dadar, M.; Pen, J.J.; Chirumbolo, S.; Aaseth, J. Chronic fatigue syndrome (CFS): Suggestions for a nutritional treatment in the therapeutic approach. Biomed. Pharmacother. 2019, 109, 1000–1007. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Tillema, A.; Leenaars, M.; Ritskes-Hoitinga, M. Enhancing search efficiency by means of a search filter for finding all studies on animal experimentation in PubMed. Lab. Anim. 2010, 44, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Ernest, S.; Hosack, A.; O’Brien, W.E.; Rosenblatt, D.S.; Nadeau, J.H. Homocysteine levels in A/J and C57BL/6J mice: Genetic, diet, gender, and parental effects. Physiol. Genom. 2005, 21, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Da Cunha, A.A.; Ferreira, A.G.K.; Wyse, A.T.S. Increased inflammatory markers in brain and blood of rats subjected to acute homocysteine administration. Metab. Brain Dis. 2010, 25, 199–206. [Google Scholar] [CrossRef]
- Algaidi, S.A.; Christie, L.A.; Jenkinson, A.M.; Whalley, L.; Riedel, G.; Platt, B. Long-term homocysteine exposure induces alterations in spatial learning, hippocampal signalling and synaptic plasticity. Exp. Neurol. 2006, 197, 8–21. [Google Scholar] [CrossRef]
- Bernardo, A.; McCord, M.; Troen, A.M.; Allison, J.D.; McDonald, M.P. Impaired spatial memory in APP-overexpressing mice on a homocysteinemia-inducing diet. Neurobiol. Aging 2007, 28, 1195–1205. [Google Scholar] [CrossRef]
- Zhuo, J.; Praticò, D. Normalization of hyperhomocysteinemia improves cognitive deficits and ameliorates brain amyloidosis of a transgenic mouse model of Alzheimer’s disease. FASEB J. 2010, 24, 3895–3902. [Google Scholar] [CrossRef] [Green Version]
- Bolivar, V.J. Intrasession and intersession habituation in mice: From inbred strain variability to linkage analysis. Neurobiol. Learn. Mem. 2009, 92, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Alzoubi, K.H.; Aburashed, Z.O.; Mayyas, F. Edaravone protects from memory impairment induced by chronic L-methionine administration. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1221–1228. [Google Scholar] [CrossRef]
- Pourié, G.; Martin, N.; Daval, J.-L.; Alberto, J.-M.; Umoret, R.; Guéant, J.-L.; Bossenmeyer-Pourié, C. The Stimulation of Neurogenesis Improves the Cognitive Status of Aging Rats Subjected to Gestational and Perinatal Deficiency of B9–12 Vitamins. Int. J. Mol. Sci. 2020, 21, 8008. [Google Scholar] [CrossRef]
- Braun, D.J.; Dimayuga, E.; Morganti, J.M.; Van Eldik, L.J. Microglial-associated responses to comorbid amyloid pathology and hyperhomocysteinemia in an aged knock-in mouse model of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 274. [Google Scholar] [CrossRef]
- Kovalska, M.; Hnilicova, P.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Effect of Methionine Diet on Time-Related Metabolic and Histopathological Changes of Rat Hippocampus in the Model of Global Brain Ischemia. Biomolecules 2020, 10, 1128. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, H.; Wang, X.; Xie, J.; An, D.; Wan, L.; Wang, J.; Zeng, Y.; Shu, X.; Westermarck, J.; et al. Direct Activation of Protein Phosphatase 2A (PP2A) by Tricyclic Sulfonamides Ameliorates Alzheimer’s Disease Pathogenesis in Cell and Animal Models. Neurotherapeutics 2020, 17, 1087–1103. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Winslow, W.; Dave, N.; Piras, I.S.; Naymik, M.; Huentelman, M.J.; Tran, A.; Caccamo, A.; Oddo, S. Maternal choline supplementation ameliorates Alzheimer’s disease pathology by reducing brain homocysteine levels across multiple generations. Mol. Psychiatry 2020, 25, 2620–2629. [Google Scholar] [CrossRef]
- Kovalska, M.; Hnilicova, P.; Kalenska, D.; Tothova, B.; Adamkov, M.; Lehotsky, J. Effect of Methionine Diet on Metabolic and Histopathological Changes of Rat Hippocampus. Int. J. Mol. Sci. 2019, 20, 6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weekman, E.M.; Sudduth, T.L.; Price, B.R.; Woolums, A.E.; Hawthorne, D.; Seaks, C.E.; Wilcock, D.M. Time course of neuropathological events in hyperhomocysteinemic amyloid depositing mice reveals early neuroinflammatory changes that precede amyloid changes and cerebrovascular events. J. Neuroinflamm. 2019, 16, 284. [Google Scholar] [CrossRef] [PubMed]
- Majtan, T.; Park, I.; Cox, A.; Branchford, B.R.; Paola, J.; Bublil, E.M.; Kraus, J.P. Behavior, body composition, and vascular phenotype of homocystinuric mice on methionine-restricted diet or enzyme replacement therapy. FASEB J. 2019, 33, 12477–12486. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.J.; Abner, E.; Bakshi, V.; Goulding, D.S.; Grau, E.M.; Lin, A.-L.; Norris, C.M.; Sudduth, T.L.; Webster, S.J.; Wilcock, D.M.; et al. Blood Flow Deficits and Cerebrovascular Changes in a Dietary Model of Hyperhomocysteinemia. ASN Neuro 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Deng, J.; Shen, Y.; Zhang, P.; Dong, H.; Xie, Z.; Xiong, L. Hyperhomocysteinemia is key for increased susceptibility to PND in aged mice. Ann. Clin. Transl. Neurol. 2019, 6, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.A.; Raizman, M.; Weizmann, N.; Wasek, B.; Arning, E.; Bottiglieri, T.; Tirosh, O.; Troen, A.M. Betaine attenuates pathology by stimulating lipid oxidation in liver and regulating phospholipid metabolism in brain of methionine-choline–deficient rats. FASEB J. 2019, 33, 9334–9349. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.J.; Bachstetter, A.D.; Sudduth, T.L.; Wilcock, D.M.; Watterson, D.M.; Van Eldik, L.J. Genetic knockout of myosin light chain kinase (MLCK210) prevents cerebral microhemorrhages and attenuates neuroinflammation in a mouse model of vascular cognitive impairment and dementia. GeroScience 2019, 41, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Bahous, R.H.; Cosín-Tomás, M.; Deng, L.; Leclerc, D.; Malysheva, O.; Ho, M.-K.; Pallàs, M.; Kaliman, P.; Bedell, B.J.; Caudill, M.A.; et al. Early Manifestations of Brain Aging in Mice Due to Low Dietary Folate and Mild MTHFR Deficiency. Mol. Neurobiol. 2019, 56, 4175–4191. [Google Scholar] [CrossRef]
- Geoffroy, A.; Saber-Cherif, L.; Pourié, G.; Helle, D.; Umoret, R.; Guéant, J.-L.; Bossenmeyer-Pourié, C.; Daval, J.-L. Developmental Impairments in a Rat Model of Methyl Donor Deficiency: Effects of a Late Maternal Supplementation with Folic Acid. Int. J. Mol. Sci. 2019, 20, 973. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ni, S.; Li, Q.; Wang, J.-Z.; Yang, Y. Folate/Vitamin B Alleviates Hyperhomocysteinemia-Induced Alzheimer-Like Pathologies in Rat Retina. Neurosci. Bull. 2019, 35, 325–335. [Google Scholar] [CrossRef]
- Mijailovic, N.; Selakovic, D.; Joksimovic, J.; Mihailovic, V.; Katanic, J.; Jakovljevic, V.; Nikolic, T.; Bolevich, S.; Zivkovic, V.; Pantic, M.; et al. The anxiolytic effects of atorvastatin and simvastatin on dietary-induced increase in homocysteine levels in rats. Mol. Cell. Biochem. 2019, 452, 199–217. [Google Scholar] [CrossRef]
- Di Meco, A.; Li, J.-G.; Barrero, C.; Merali, S.; Praticò, D. Elevated levels of brain homocysteine directly modulate the pathological phenotype of a mouse model of tauopathy. Mol. Psychiatry 2019, 24, 1696–1706. [Google Scholar] [CrossRef]
- Mahaman, Y.A.R.; Huang, F.; Wu, M.; Wang, Y.; Wei, Z.; Bao, J.; Salissou, M.T.M.; Ke, D.; Wang, Q.; Liu, R.; et al. Moringa Oleifera Alleviates Homocysteine-Induced Alzheimer’s Disease-Like Pathology and Cognitive Impairments. J. Alzheimer’s Dis. 2018, 63, 1141–1159. [Google Scholar] [CrossRef] [Green Version]
- Nuru, M.; Muradashvili, N.; Kalani, A.; Lominadze, D.; Tyagi, N. High methionine, low folate and low vitamin B6/B12 (HM-LF-LV) diet causes neurodegeneration and subsequent short-term memory loss. Metab. Brain Dis. 2018, 33, 1923–1934. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Mhaidat, N.M.; Obaid, E.A.; Khabour, O.F. Caffeine Prevents Memory Impairment Induced by Hyperhomocysteinemia. J. Mol. Neurosci. 2018, 66, 222–228. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, M.M.; Yuan, L.; Huang, L.L.; Liao, J.H.; Yu, X.L.; Su, C.; Chen, Y.H.; Yang, Y.Y.; Yu, H.; et al. Chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice. PLoS ONE 2018, 13, e0202910. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Li, S.; Yan, J.; Wilson, J.X.; Huang, G. Maternal Folic Acid Supplementation During Pregnancy Improves Neurobehavioral Development in Rat Offspring. Mol. Neurobiol. 2018, 55, 2676–2684. [Google Scholar] [CrossRef]
- Zeng, K.; Li, M.; Hu, J.; Mahaman, Y.A.R.; Bao, J.; Huang, F.; Xia, Y.; Liu, X.; Wang, Q.; Wang, J.-Z.; et al. Ginkgo biloba Extract EGb761 Attenuates Hyperhomocysteinemia-induced AD Like Tau Hyperphosphorylation and Cognitive Impairment in Rats. Curr. Alzheimer Res. 2018, 15, 89–99. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Neuroprotective Effect of Hydrogen Sulfide in Hyperhomocysteinemia Is Mediated Through Antioxidant Action Involving Nrf2. NeuroMol. Med. 2018, 20, 475–490. [Google Scholar] [CrossRef]
- Kolling, J.; Longoni, A.; Siebert, C.; dos Santos, T.M.; Marques, E.P.; Carletti, J.; Pereira, L.O.; Wyse, A.T.S. Severe Hyperhomocysteinemia Decreases Creatine Kinase Activity and Causes Memory Impairment: Neuroprotective Role of Creatine. Neurotox. Res. 2017, 32, 585–593. [Google Scholar] [CrossRef]
- Kumar, M.; Modi, M.; Sandhir, R. Hydrogen sulfide attenuates homocysteine-induced cognitive deficits and neurochemical alterations by improving endogenous hydrogen sulfide levels. BioFactors 2017, 43, 434–450. [Google Scholar] [CrossRef]
- Li, J.-G.; Barrero, C.; Merali, S.; Praticò, D. Genetic absence of ALOX5 protects from homocysteine-induced memory impairment, tau phosphorylation and synaptic pathology. Hum. Mol. Genet. 2017, 26, 1855–1862. [Google Scholar] [CrossRef]
- Li, J.-G.; Barrero, C.; Merali, S.; Praticò, D. Five lipoxygenase hypomethylation mediates the homocysteine effect on Alzheimer’s phenotype. Sci. Rep. 2017, 7, 46002. [Google Scholar] [CrossRef] [Green Version]
- Hemanth Kumar, B.; Arun Reddy, R.; Mahesh Kumar, J.; Dinesh Kumar, B.; Diwan, P.V. Effects of fisetin on hyperhomocysteinemia-induced experimental endothelial dysfunction and vascular dementia. Can. J. Physiol. Pharmacol. 2017, 95, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Hemanth Kumar, B.; Dinesh Kumar, B.; Diwan, P.V. Hesperidin, a citrus flavonoid, protects against l-methionine-induced hyperhomocysteinemia by abrogation of oxidative stress, endothelial dysfunction and neurotoxicity in Wistar rats. Pharm. Biol. 2017, 55, 146–155. [Google Scholar] [CrossRef]
- Li, M.; Zhang, P.; Wei, H.; Li, M.-H.; Zou, W.; Li, X.; Gu, H.-F.; Tang, X.-Q. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cognitive Dysfunction by Inhibition of Reactive Aldehydes Involving Upregulation of ALDH2. Int. J. Neuropsychopharmacol. 2016, 20, pyw103. [Google Scholar] [CrossRef]
- Weekman, E.M.; Sudduth, T.L.; Caverly, C.N.; Kopper, T.J.; Phillips, O.W.; Powell, D.K.; Wilcock, D.M. Reduced Efficacy of Anti-Aß Immunotherapy in a Mouse Model of Amyloid Deposition and Vascular Cognitive Impairment Comorbidity. J. Neurosci. 2016, 36, 9896–9907. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.R.; Mabasa, L.; Crane, C.; Park, C.S.; Venâncio, V.P.; Bianchi, M.L.P.; Antunes, L.M.G. Maternal vitamin B 6 deficient or supplemented diets on expression of genes related to GABAergic, serotonergic, or glutamatergic pathways in hippocampus of rat dams and their offspring. Mol. Nutr. Food Res. 2016, 60, 1615–1624. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kyles, P.; Kalani, A.; Tyagi, N. Hydrogen Sulfide Ameliorates Homocysteine-Induced Alzheimer’s Disease-Like Pathology, Blood–Brain Barrier Disruption, and Synaptic Disorder. Mol. Neurobiol. 2016, 53, 2451–2467. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kruyer, A.; Yao, Y.; Feierman, E.; Richards, A.; Strickland, S.; Norris, E.H. Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the β-amyloid fibrinogen interaction. J. Thromb. Haemost. 2016, 14, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Modi, K.K.; Roy, A.; Brahmachari, S.; Rangasamy, S.B.; Pahan, K. Cinnamon and Its Metabolite Sodium Benzoate Attenuate the Activation of p21rac and Protect Memory and Learning in an Animal Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0130398. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Sugimoto, Y.; Fujita, A.; Kanouchi, H. Ethanol extract of Brazilian propolis ameliorates cognitive dysfunction and suppressed protein aggregations caused by hyperhomocysteinemia. Biosci. Biotechnol. Biochem. 2015, 79, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Kunisawa, K.; Nakashima, N.; Nagao, M.; Nomura, T.; Kinoshita, S.; Hiramatsu, M. Betaine prevents homocysteine-induced memory impairment via matrix metalloproteinase-9 in the frontal cortex. Behav. Brain Res. 2015, 292, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Jadavji, N.M.; Deng, L.; Malysheva, O.; Caudill, M.A.; Rozen, R. MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short-term memory and increased apoptosis in the hippocampus of wild-type offspring. Neuroscience 2015, 300, 1–9. [Google Scholar] [CrossRef]
- Zhao, H.; Ji, Z.-H.; Liu, C.; Yu, X.-Y. Neuroprotection and mechanisms of atractylenolide III in preventing learning and memory impairment induced by chronic high-dose homocysteine administration in rats. Neuroscience 2015, 290, 485–491. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Farr, T.D.; Lips, J.; Khalil, A.A.; Boehm-Sturm, P.; Foddis, M.; Harms, C.; Füchtemeier, M.; Dirnagl, U. Elevated levels of plasma homocysteine, deficiencies in dietary folic acid and uracil–DNA glycosylase impair learning in a mouse model of vascular cognitive impairment. Behav. Brain Res. 2015, 283, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Jing, M.; Rech, L.; Wu, Y.; Goltz, D.; Taylor, C.G.; House, J.D. Effects of zinc deficiency and zinc supplementation on homocysteine levels and related enzyme expression in rats. J. Trace Elem. Med. Biol. 2015, 30, 77–82. [Google Scholar] [CrossRef]
- Jiang, X.; Chai, G.-S.; Wang, Z.-H.; Hu, Y.; Li, X.-G.; Ma, Z.-W.; Wang, Q.; Wang, J.-Z.; Liu, G.-P. CaMKII-dependent dendrite ramification and spine generation promote spatial training-induced memory improvement in a rat model of sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 867–876. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, R.; Chen, R.; Tian, Q.; Zeng, K.; Hu, J.; Liu, X.; Wang, Q.; Wang, P.; Wang, X.-C.; et al. Novel Multipotent AChEI-CCB Attenuates Hyperhomocysteinemia-Induced Memory Deficits and Neuropathologies in Rats. J. Alzheimer’s Dis. 2014, 42, 1029–1039. [Google Scholar] [CrossRef]
- Berrocal-Zaragoza, M.I.; Sequeira, J.M.; Murphy, M.M.; Fernandez-Ballart, J.D.; Abdel Baki, S.G.; Bergold, P.J.; Quadros, E.V. Folate deficiency in rat pups during weaning causes learning and memory deficits. Br. J. Nutr. 2014, 112, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- De Rezende, M.M.; D’Almeida, V. Central and Systemic Responses to Methionine-Induced Hyperhomocysteinemia in Mice. PLoS ONE 2014, 9, e105704. [Google Scholar] [CrossRef] [Green Version]
- Muradashvili, N.; Tyagi, R.; Metreveli, N.; Tyagi, S.C.; Lominadze, D. Ablation of MMP9 Gene Ameliorates Paracellular Permeability and Fibrinogen–Amyloid Beta Complex Formation during Hyperhomocysteinemia. J. Cereb. Blood Flow Metab. 2014, 34, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-H.; Tang, J.-P.; Zhang, P.; Li, X.; Wang, C.-Y.; Wei, H.-J.; Yang, X.-F.; Zou, W.; Tang, X.-Q. Disturbance of endogenous hydrogen sulfide generation and endoplasmic reticulum stress in hippocampus are involved in homocysteine-induced defect in learning and memory of rats. Behav. Brain Res. 2014, 262, 35–41. [Google Scholar] [CrossRef]
- Li, J.-G.; Chu, J.; Barrero, C.; Merali, S.; Praticò, D. Homocysteine exacerbates β-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann. Neurol. 2014, 75, 851–863. [Google Scholar] [CrossRef]
- Lu, Y.-Q.; Luo, Y.; He, Z.-F.; Chen, J.; Yan, B.; Wang, Y.; Yu, Q. Hydroxysafflor Yellow A Ameliorates Homocysteine-Induced Alzheimer-Like Pathologic Dysfunction and Memory/Synaptic Disorder. Rejuvenation Res. 2013, 16, 446–452. [Google Scholar] [CrossRef]
- Farkas, M.; Keskitalo, S.; Smith, D.E.C.; Bain, N.; Semmler, A.; Ineichen, B.; Smulders, Y.; Blom, H.; Kulic, L.; Linnebank, M. Hyperhomocysteinemia in Alzheimer’s Disease: The Hen and the Egg? J. Alzheimer’s Dis. 2013, 33, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of Hyperhomocysteinemia Models Vascular Dementia by Induction of Cerebral Microhemorrhages and Neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Chai, G.-S.; Jiang, X.; Ni, Z.-F.; Ma, Z.-W.; Xie, A.-J.; Cheng, X.-S.; Wang, Q.; Wang, J.-Z.; Liu, G.-P. Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J. Neurochem. 2013, 124, 388–396. [Google Scholar] [CrossRef]
- Koz, S.T.; Etem, E.O.; Baydas, G.; Yuce, H.; Ozercan, H.I.; Kuloğlu, T.; Koz, S.; Etem, A.; Demir, N. Effects of resveratrol on blood homocysteine level, on homocysteine induced oxidative stress, apoptosis and cognitive dysfunctions in rats. Brain Res. 2012, 1484, 29–38. [Google Scholar] [CrossRef]
- Ehrlich, D.; Humpel, C. Chronic vascular risk factors (cholesterol, homocysteine, ethanol) impair spatial memory, decline cholinergic neurons and induce blood–brain barrier leakage in rats in vivo. J. Neurol. Sci. 2012, 322, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Singh, N. Salutary effect of NFκB inhibitor and folacin in hyperhomocysteinemia–hyperlipidemia induced vascular dementia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 207–215. [Google Scholar] [CrossRef]
- Koz, S.T.; Baydas, G.; Koz, S.; Demir, N.; Nedzvetsky, V.S. Gingko biloba Extract Inhibits Oxidative Stress and Ameliorates Impaired Glial Fibrillary Acidic Protein Expression, but Can Not Improve Spatial Learning in Offspring from Hyperhomocysteinemic Rat Dams. Phyther. Res. 2012, 26, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zeng, X.-N.; Guo, H.-M.; Wu, X.-M.; Chen, H.-J.; Di, R.-K.; Wu, Y. Cognitive and neurochemical alterations in hyperhomocysteinemic rat. Neurol. Sci. 2012, 33, 39–43. [Google Scholar] [CrossRef]
- Viggiano, A.; Viggiano, E.; Monda, M.; Ingrosso, D.; Perna, A.F.; De Luca, B. Methionine-enriched diet decreases hippocampal antioxidant defences and impairs spontaneous behaviour and long-term potentiation in rats. Brain Res. 2012, 1471, 66–74. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Ricceri, L.; Cavallaro, R.A.; Isopi, E.; Mangia, F.; Fiorenza, M.T.; Scarpa, S. S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice. Neurobiol. Aging 2012, 33, 1482.e1–1482.e16. [Google Scholar] [CrossRef]
- Sain, H.; Sharma, B.; Jaggi, A.S.; Singh, N. Pharmacological investigations on potential of peroxisome proliferator-activated receptor-gamma agonists in hyperhomocysteinemia-induced vascular dementia in rats. Neuroscience 2011, 192, 322–333. [Google Scholar] [CrossRef]
- McCampbell, A.; Wessner, K.; Marlatt, M.W.; Wolffe, C.; Toolan, D.; Podtelezhnikov, A.; Yeh, S.; Zhang, R.; Szczerba, P.; Tanis, K.Q.; et al. Induction of Alzheimer’s-like changes in brain of mice expressing mutant APP fed excess methionine. J. Neurochem. 2011, 116, 82–92. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, Z.; Zhao, N.; Liu, D.; Guo, Z.-Y.; Tan, L.; Hu, J.; Wang, Q.; Wang, J.-Z.; Zhu, L.-Q. Acetyl- L-Carnitine Attenuates Homocysteine-Induced Alzheimer-Like Histopathological and Behavioral Abnormalities. Rejuvenation Res. 2011, 14, 669–679. [Google Scholar] [CrossRef]
- Vijayanathan, V.; Gulinello, M.; Ali, N.; Cole, P.D. Persistent cognitive deficits, induced by intrathecal methotrexate, are associated with elevated CSF concentrations of excitotoxic glutamate analogs and can be reversed by an NMDA antagonist. Behav. Brain Res. 2011, 225, 491–497. [Google Scholar] [CrossRef]
- Naim, M.Y.; Friess, S.; Smith, C.; Ralston, J.; Ryall, K.; Helfaer, M.A.; Margulies, S.S. Folic Acid Enhances Early Functional Recovery in a Piglet Model of Pediatric Head Injury. Dev. Neurosci. 2010, 32, 454–467. [Google Scholar] [CrossRef] [Green Version]
- Ataie, A.; Sabetkasaei, M.; Haghparast, A.; Moghaddam, A.H.; Kazeminejad, B. Neuroprotective effects of the polyphenolic antioxidant agent, Curcumin, against homocysteine-induced cognitive impairment and oxidative stress in the rat. Pharmacol. Biochem. Behav. 2010, 96, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Evola, M.; Hall, A.; Wall, T.; Young, A.; Grammas, P. Oxidative stress impairs learning and memory in apoE knockout mice. Pharmacol. Biochem. Behav. 2010, 96, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calegare, B.F.A.; Fernandes, L.; Tufik, S.; D’Almeida, V. Biochemical, biometrical and behavioral changes in male offspring of sleep-deprived mice. Psychoneuroendocrinology 2010, 35, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.-M.; Praticò, D. Severe In Vivo Hyper-Homocysteinemia is not Associated with Elevation of Amyloid-β Peptides in the Tg2576 Mice. J. Alzheimer’s Dis. 2010, 21, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.-M.; Praticò, D. Acceleration of brain amyloidosis in an Alzheimer’s disease mouse model by a folate, vitamin B6 and B12-deficient diet. Exp. Gerontol. 2010, 45, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.-M.; Portugal, G.; Kruger, W.; Wang, H.; Gould, T.; Pratico, D. Diet-Induced Hyperhomocysteinemia Increases Amyloid-β Formation and Deposition in a Mouse Model of Alzheimers Disease. Curr. Alzheimer Res. 2010, 7, 140–149. [Google Scholar] [CrossRef]
- Matté, C.; Pereira, L.O.; Dos Santos, T.M.; Mackedanz, V.; Cunha, A.A.; Netto, C.A.; Wyse, A.T.S. Acute homocysteine administration impairs memory consolidation on inhibitory avoidance task and decreases hippocampal brain-derived neurotrophic factor immunocontent: Prevention by folic acid treatment. Neuroscience 2009, 163, 1039–1045. [Google Scholar] [CrossRef]
- Blaise, S.A.; Nédélec, E.; Alberto, J.-M.; Schroeder, H.; Audonnet, S.; Bossenmeyer-Pourié, C.; Guéant, J.-L.; Daval, J.-L. Short hypoxia could attenuate the adverse effects of hyperhomocysteinemia on the developing rat brain by inducing neurogenesis. Exp. Neurol. 2009, 216, 231–238. [Google Scholar] [CrossRef]
- Zhang, C.-E.; Wei, W.; Liu, Y.-H.; Peng, J.-H.; Tian, Q.; Liu, G.-P.; Zhang, Y.; Wang, J.-Z. Hyperhomocysteinemia Increases β-Amyloid by Enhancing Expression of γ-Secretase and Phosphorylation of Amyloid Precursor Protein in Rat Brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, G.; Harms, C.; Sobol, R.W.; Cardozo-Pelaez, F.; Linhart, H.; Winter, B.; Balkaya, M.; Gertz, K.; Gay, S.B.; Cox, D.; et al. Folate Deficiency Induces Neurodegeneration and Brain Dysfunction in Mice Lacking Uracil DNA Glycosylase. J. Neurosci. 2008, 28, 7219–7230. [Google Scholar] [CrossRef]
- Baydas, G.; Koz, S.T.; Tuzcu, M.; Nedzvetsky, V.S. Melatonin prevents gestational hyperhomocysteinemia-associated alterations in neurobehavioral developments in rats. J. Pineal Res. 2008, 44, 181–188. [Google Scholar] [CrossRef]
- Troen, A.M.; Chao, W.-H.; Crivello, N.A.; D’Anci, K.E.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. Cognitive Impairment in Folate-Deficient Rats Corresponds to Depleted Brain Phosphatidylcholine and Is Prevented by Dietary Methionine without Lowering Plasma Homocysteine. J. Nutr. 2008, 138, 2502–2509. [Google Scholar] [CrossRef] [Green Version]
- Troen, A.M.; Shea-Budgell, M.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12474–12479. [Google Scholar] [CrossRef] [Green Version]
- Lalonde, R.; Barraud, H.; Ravey, J.; Guéant, J.-L.; Bronowicki, J.-P.; Strazielle, C. Effects of a B-vitamin-deficient diet on exploratory activity, motor coordination, and spatial learning in young adult Balb/c mice. Brain Res. 2008, 1188, 122–131. [Google Scholar] [CrossRef]
- Zhang, C.-E.; Tian, Q.; Wei, W.; Peng, J.-H.; Liu, G.-P.; Zhou, X.-W.; Wang, Q.; Wang, D.-W.; Wang, J.-Z. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef]
- Baydas, G.; Koz, S.T.; Tuzcu, M.; Nedzvetsky, V.S.; Etem, E. Effects of maternal hyperhomocysteinemia induced by high methionine diet on the learning and memory performance in offspring. Int. J. Dev. Neurosci. 2007, 25, 133–139. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Rodriguez de Turco, E.B.; DeRosa, S.; Howard, A.; Cruz-Sanchez, F.; Sambamurti, K.; Refolo, L.; Petanceska, S.; Pappolla, M.A. Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid β peptide levels. Neurobiol. Dis. 2006, 22, 651–656. [Google Scholar] [CrossRef]
- Baydas, G.; Özer, M.; Yasar, A.; Tuzcu, M.; Koz, S.T. Melatonin improves learning and memory performances impaired by hyperhomocysteinemia in rats. Brain Res. 2005, 1046, 187–194. [Google Scholar] [CrossRef]
- Robert, K.; Santiard-Baron, D.; Chassé, J.-F.; Paly, E.; Aupetit, J.; Kamoun, P.; London, J.; Janel, N. The neuronal SAPK/JNK pathway is altered in a murine model of hyperhomocysteinemia. J. Neurochem. 2004, 89, 33–43. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.J.; Kim, J.; Chang, N. Effects of dietary folic acid supplementation on cerebrovascular endothelial dysfunction in rats with induced hyperhomocysteinemia. Brain Res. 2004, 996, 139–147. [Google Scholar] [CrossRef]
- Streck, E.L.; Bavaresco, C.S.; Netto, C.A.; Wyse, A.T.D.S. Chronic hyperhomocysteinemia provokes a memory deficit in rats in the Morris water maze task. Behav. Brain Res. 2004, 153, 377–381. [Google Scholar] [CrossRef]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic Acid Deficiency and Homocysteine Impair DNA Repair in Hippocampal Neurons and Sensitize Them to Amyloid Toxicity in Experimental Models of Alzheimer’s Disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Karaplis, A.C.; Ackerman, S.L.; Pogribny, I.P.; Melnyk, S.; Lussier-Cacan, S.; Chen, M.F.; Pai, A.; John, S.W.; Smith, R.S.; et al. Mice deficient in methylenetetrahydrofolate reductase exhibit hyperhomocysteinemia and decreased methylation capacity, with neuropathology and aortic lipid deposition. Hum. Mol. Genet. 2001, 10, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Gospe, S.M.; Gietzen, D.W.; Summers, P.J.; Lunetta, J.M.; Miller, J.W.; Selhub, J.; Ellis, W.G.; Clifford, A.J. Behavioral and neurochemical changes in folate-deficient mice. Physiol. Behav. 1995, 58, 935–941. [Google Scholar] [CrossRef]
- Huang, F.; Chen, X.; Jiang, X.; Niu, J.; Cui, C.; Chen, Z.; Sun, J. Betaine ameliorates prenatal valproic-acid-induced autism-like behavioral abnormalities in mice by promoting homocysteine metabolism. Psychiatry Clin. Neurosci. 2019, 73, 317–322. [Google Scholar] [CrossRef]
- Gupta, S.; Wang, L.; Kruger, W.D. Betaine supplementation is less effective than methionine restriction in correcting phenotypes of CBS deficient mice. J. Inherit. Metab. Dis. 2016, 39, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Kakimoto, T.; Otsuka, A.; Kawaguchi, H.; Ogata, K.; Tanimoto, A.; Kanouchi, H. Plasma homocysteine concentrations in novel microminipigs. In Vivo 2014, 28, 579–582. [Google Scholar]
- Liu, Y.; Liu, Y.-Q.; Morita, T.; Sugiyama, K. Effects of Betaine Supplementation and Choline Deficiency on Folate Deficiency-Induced Hyperhomocysteinemia in Rats. J. Nutr. Sci. Vitaminol. 2012, 58, 69–77. [Google Scholar] [CrossRef]
- Gupta, S.; Kruger, W.D. Cystathionine Beta-Synthase Deficiency Causes Fat Loss in Mice. PLoS ONE 2011, 6, e27598. [Google Scholar] [CrossRef]
- Mikael, L.G.; Wang, X.-L.; Wu, Q.; Jiang, H.; Maclean, K.N.; Rozen, R. Hyperhomocysteinemia is associated with hypertriglyceridemia in mice with methylenetetrahydrofolate reductase deficiency. Mol. Genet. Metab. 2009, 98, 187–194. [Google Scholar] [CrossRef]
- Setoue, M.; Ohuchi, S.; Morita, T.; Sugiyama, K. Hyperhomocysteinemia Induced by Guanidinoacetic Acid Is Effectively Suppressed by Choline and Betaine in Rats. Biosci. Biotechnol. Biochem. 2008, 72, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burren, K.A.; Savery, D.; Massa, V.; Kok, R.M.; Scott, J.M.; Blom, H.J.; Copp, A.J.; Greene, N.D.E. Gene-environment interactions in the causation of neural tube defects: Folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum. Mol. Genet. 2008, 17, 3675–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chwatko, G.; Boers, G.H.J.; Strauss, K.A.; Shih, D.M.; Jakubowski, H. Mutations in methylenetetrahydrofolate reductase or cystathionine β-syntase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007, 21, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Jiang, X.; Yang, F.; Li, Z.; Liao, D.; Trial, J.; Magera, M.; Durante, W.; Yang, X.; Wang, H. Hyperhomocysteinemia inhibits post-injury reendothelialization in mice. Cardiovasc. Res. 2006, 69, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J.; Dole, V.S.; Loscalzo, J.; Handy, D.E.; Wagner, D.D. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [Green Version]
- Sauls, D.L.; Boyd, L.C.; Allen, J.C.; Hoffman, M. Differences in the metabolic response to exogenous homocysteine in juvenile and adult rabbits. J. Nutr. Biochem. 2004, 15, 96–102. [Google Scholar] [CrossRef]
- Wang, L.; Jhee, K.-H.; Hua, X.; DiBello, P.M.; Jacobsen, D.W.; Kruger, W.D. Modulation of Cystathionine β-Synthase Level Regulates Total Serum Homocysteine in Mice. Circ. Res. 2004, 94, 1318–1324. [Google Scholar] [CrossRef] [Green Version]
- Neves, M.F.; Endemann, D.; Amiri, F.; Virdis, A.; Pu, Q.; Rozen, R.; Schiffrin, E.L. Small artery mechanics in hyperhomocysteinemic mice. J. Hypertens. 2004, 22, 959–966. [Google Scholar] [CrossRef]
- Hossain, G.S.; van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; de Koning, A.B.L.; Tang, D.; Wu, D.; Falk, E.; et al. TDAG51 Is Induced by Homocysteine, Promotes Detachment-mediated Programmed Cell Death, and Contributes to the Development of Atherosclerosis in Hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, H.; Chang, N. Hyperhomocysteinemia Due to Short-Term Folate Deprivation Is Related to Electron Microscopic Changes in the Rat Brain. J. Nutr. 2002, 132, 3418–3421. [Google Scholar] [CrossRef] [Green Version]
- Streck, E.L.; Matte, C.; Vieira, P.S.; Rombaldi, F.; Wannmacher, C.M.D.; Wajner, M.; Wyse, A.T.S. Reduction of Na+, K+-ATPase activity in hippocampus of rats subjected to chemically induced hyperhomocysteinemia. Neurochem. Res. 2002, 27, 1593–1598. [Google Scholar] [CrossRef]
- Baumbach, G.L.; Sigmund, C.D.; Bottiglieri, T.; Lentz, S.R. Structure of Cerebral Arterioles in Cystathionine β-Synthase-Deficient Mice. Circ. Res. 2002, 91, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Dayal, S.; Bottiglieri, T.; Arning, E.; Maeda, N.; Malinow, M.R.; Sigmund, C.D.; Heistad, D.D.; Faraci, F.M.; Lentz, S.R. Endothelial Dysfunction and Elevation of S -Adenosylhomocysteine in Cystathionine β-Synthase–Deficient Mice. Circ. Res. 2001, 88, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Lentz, S.R.; Sobey, C.G.; Piegors, D.J.; Bhopatkar, M.Y.; Faraci, F.M.; Malinow, M.R.; Heistad, D.D. Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Osada, J.; Aratani, Y.; Kluckman, K.; Reddick, R.; Malinow, M.R.; Maeda, N. Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 1995, 92, 1585–1589. [Google Scholar] [CrossRef] [Green Version]
- Harker, L.A.; Harlan, J.M.; Ross, R. Effect of sulfinpyrazone on homocysteine-induced endothelial injury and arteriosclerosis in baboons. Circ. Res. 1983, 53, 731–739. [Google Scholar] [CrossRef] [Green Version]







|
|
|
|
|
|
|
|
| Vessels: |
| - Arteries: intimal thickening, media destruction, fibrous plaques, thrombosis |
| → cf.: atherosclerosis and its complications |
| - Veins: deep leg vein thrombosis, embolism |
| → cf. thrombosis, embolism |
| If left untreated, most patients die in childhood or adolescence from consequences of vascular damage: arterial and venous thrombosis, embolism, myocardial infarction and stroke |
| Central nervous system: |
| - Mental retardation, epilepsy |
| → cf. cognitive impairment, dementia, depression |
| Skeleton: |
| - Marfanoid habit with arachnodactyly, bone deformities, osteoporosis |
| → cf. increased fracture rate |
| Eyes: |
| - Lens dislocation and severe myopia, also possible cataract, optic atrophy and retinal degeneration |
| → cf. retinopathies and macular degeneration |
| Hypo-methylation—Rev [52]; also see Figure 3: HCys ↑ → SAM/SAH ↓ → hypo-methylation of the presenilin 1 gene → increased β-amyloid formation. Hypo-methylation of the enzyme protein phosphatase 2A → loss of activity for phosphate cleavage of protein tau → accumulation of over-phosphorylated protein tau in neurofibrils → deposition of neurofibrillary tangles. | Clinical studies—Rev [73,74,75]: Plasma HCys negatively correlates with the thickness of the medial, inferior temporal lobe in normal subjects; equally in Alzheimer’s patients (already lower baseline values). Meta-analysis (77 case-control studies, 33 prospective studies, 46,000 subjects): | |
| Neurotoxicity—Rev [73]; also see Figure 4E,F: HCys and oxidation products (homocysteic acid) activate NMDA receptors → excitotoxicity (cellular Ca2+ increase → activation of proteases and radical formation → cell death = neuronal degeneration). Increased formation of ROS → activation of NFκB → inflammatory reaction. | Plasma-HCys | Risk |
| ≥15 μM | 3-fold for cognitive impairment | |
| ≥14 μM | 2-fold for Alzheimer’s dementia | |
| Of approx. 10 placebo-controlled intervention studies with the three B-vitamins (B6, B12, folate), only five meet the decisive criteria: primary preventive approach, increased HCys starting level, study duration of at least two years, adequate vitamin dosage, proven decline in cognitive parameters in the placebo group. Significant results of these studies in favor of the vitamins: reduction of the brain atrophy rate, mainly gray matter, significantly better values for dementia status, MMSE (mini mental state evaluation) and learning test. Positive influence of plasma omega-3 fatty acid level on the effect of the B-vitamins [76]. Patients in the Alzheimer’s prodromal stage benefit from multi-nutrients with B-vitamins and omega-3 fatty acids: significantly better dementia status. The effect correlates directly with the baseline MMSE value [77] → importance of early start of prevention! | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieraad, H.; Pannwitz, N.; Bruin, N.d.; Geisslinger, G.; Till, U. Hyperhomocysteinemia: Metabolic Role and Animal Studies with a Focus on Cognitive Performance and Decline—A Review. Biomolecules 2021, 11, 1546. https://doi.org/10.3390/biom11101546
Nieraad H, Pannwitz N, Bruin Nd, Geisslinger G, Till U. Hyperhomocysteinemia: Metabolic Role and Animal Studies with a Focus on Cognitive Performance and Decline—A Review. Biomolecules. 2021; 11(10):1546. https://doi.org/10.3390/biom11101546
Chicago/Turabian StyleNieraad, Hendrik, Nina Pannwitz, Natasja de Bruin, Gerd Geisslinger, and Uwe Till. 2021. "Hyperhomocysteinemia: Metabolic Role and Animal Studies with a Focus on Cognitive Performance and Decline—A Review" Biomolecules 11, no. 10: 1546. https://doi.org/10.3390/biom11101546