Emerging Roles for the INK4a/ARF (CDKN2A) Locus in Adipose Tissue: Implications for Obesity and Type 2 Diabetes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
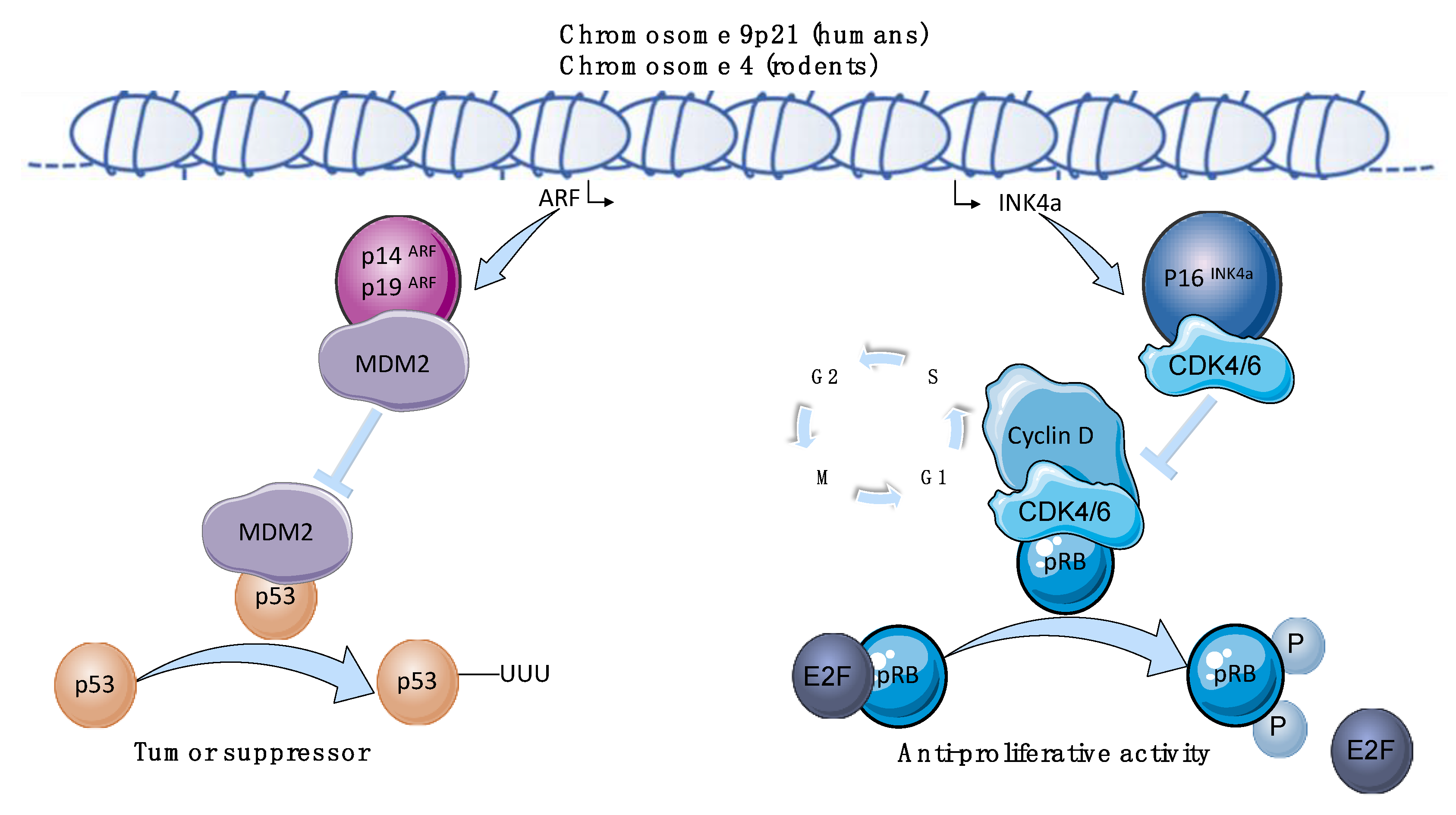
:1. Introduction
2. The INK4a/ARF Locus: A Balance between Adipogenesis and Senescence
3. The INK4a/ARF Locus: A Role in Adipocyte Insulin Sensitivity and Lipid Storage
4. The INK4a/ARF Locus: A Role in Adipose Tissue Inflammation
5. The INK4a/ARF Locus: A Role in Adipose Tissue Oxidative Activity and Browning
6. The INK4a/ARF Locus: An Emerging Key Actor in Metabolic Functions
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lam, Y.Y.; Ravussin, E. Indirect calorimetry: An indispensable tool to understand and predict obesity. Eur. J. Clin. Nutr. 2017, 71, 318–322. [Google Scholar] [CrossRef]
- González-Muniesa, P.; Mártinez-González, M.A.; Hu, F.B.; Després, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primer 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Qasim, A.; Turcotte, M.; de Souza, R.J.; Samaan, M.C.; Champredon, D.; Dushoff, J.; Speakman, J.R.; Meyre, D. On the origin of obesity: Identifying the biological, environmental and cultural drivers of genetic risk among human populations. Obes. Rev. 2018, 19, 121–149. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.C.; Jiang, Y.; Graff, J.M. Emerging Roles of Adipose Progenitor Cells in Tissue Development, Homeostasis, Expansion and Thermogenesis. Trends Endocrinol. Metab. 2016, 27, 574–585. [Google Scholar] [CrossRef]
- Lecoutre, S.; Petrus, P.; Rydén, M.; Breton, C. Transgenerational Epigenetic Mechanisms in Adipose Tissue Development. Trends Endocrinol. Metab. 2018, 29, 675–685. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocyte lineages: Tracing back the origins of fat. Biochim. Biophys. Acta 2014, 1842, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali, A.; Murano, I.; Zingaretti, M.C.; Frontini, A.; Ricquier, D.; Cinti, S. The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J. Lipid Res. 2012, 53, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, J.; Seale, P. Beige Can Be Slimming. Science 2010, 328, 1113–1114. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, C.T.; Kiefer, F.W. Adipose tissue browning in mice and humans. J. Endocrinol. 2019, 241, R97–R109. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Wouters, K.; Paumelle, R.; Staels, B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: What have we learned from GWASs? Trends Endocrinol. Metab. TEM 2015, 26, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Sharma, R.B.; Nwosu, B.U.; Alonso, L.C. Islet biology, the CDKN2A/B locus and type 2 diabetes risk. Diabetologia 2016, 59, 1579–1593. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Voight, B.F.; Teslovich, T.M.; Ferreira, T.; Segrè, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef]
- Cauchi, S.; Meyre, D.; Durand, E.; Proença, C.; Marre, M.; Hadjadj, S.; Choquet, H.; De Graeve, F.; Gaget, S.; Allegaert, F.; et al. Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS ONE 2008, 3, e2031. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.; Murray, R.; Cheong, C.; Teh, A.L.; Clarke-Harris, R.; Barton, S.; Costello, P.; Garratt, E.; Cook, E.; Titcombe, P.; et al. ANRIL Promoter DNA Methylation: A Perinatal Marker for Later Adiposity. EBioMedicine 2017, 19, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef]
- Marquez, M.P.; Alencastro, F.; Madrigal, A.; Jimenez, J.L.; Blanco, G.; Gureghian, A.; Keagy, L.; Lee, C.; Liu, R.; Tan, L.; et al. The Role of Cellular Proliferation in Adipogenic Differentiation of Human Adipose Tissue-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2017, 26, 1578–1595. [Google Scholar] [CrossRef]
- Smas, C.M.; Sul, H.S. Control of adipocyte differentiation. Biochem. J. 1995, 309, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Patel, Y.M.; Lane, M.D. Mitotic clonal expansion during preadipocyte differentiation: Calpain-mediated turnover of p27. J. Biol. Chem. 2000, 275, 17653–17660. [Google Scholar] [CrossRef] [Green Version]
- Reichert, M.; Eick, D. Analysis of cell cycle arrest in adipocyte differentiation. Oncogene 1999, 18, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional regulation of adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Siersbæk, R.; Nielsen, R.; John, S.; Sung, M.H.; Baek, S.; Loft, A.; Hager, G.L.; Mandrup, S. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J. 2011, 30, 1459–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siersbæk, R.; Baek, S.; Rabiee, A.; Nielsen, R.; Traynor, S.; Clark, N.; Sandelin, A.; Jensen, O.N.; Sung, M.H.; Hager, G.L.; et al. Molecular architecture of transcription factor hotspots in early adipogenesis. Cell Rep. 2014, 7, 1434–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siersbæk, R.; Nielsen, R.; Mandrup, S. Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol. Metab. TEM 2012, 23, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; Deleye, Y.; Hannou, S.A.; Vanhoutte, J.; Maréchal, X.; Coisne, A.; Tagzirt, M.; Derudas, B.; Bouchaert, E.; Duhem, C.; et al. The tumour suppressor CDKN2A/p16INK4a regulates adipogenesis and bone marrow-dependent development of perivascular adipose tissue. Diabetes Vasc. Dis. Res. 2017, 14, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Cho, Y.; Lee, J.; Jeong, H.; Lee, Y.; Kim, S.I.; Kim, C.H.; Lee, H.W.; Nam, K.T. Sexually dimorphic leanness and hypermobility in p16Ink4a/CDKN2A-deficient mice coincides with phenotypic changes in the cerebellum. Sci. Rep. 2019, 9, 11167. [Google Scholar] [CrossRef] [Green Version]
- Abella, A.; Dubus, P.; Malumbres, M.; Rane, S.G.; Kiyokawa, H.; Sicard, A.; Vignon, F.; Langin, D.; Barbacid, M.; Fajas, L. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab. 2005, 2, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, Y.; Li, S.; Yang, Y.; Dai, Z.; Wang, F.; Wu, Z.; Tso, P.; Wu, G. E2F1 Regulates Adipocyte Differentiation and Adipogenesis by Activating ICAT. Cells 2020, 9, 1024. [Google Scholar] [CrossRef] [Green Version]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Porse, B.T.; Pedersen, T.A.; Xu, X.; Lindberg, B.; Wewer, U.M.; Friis-Hansen, L.; Nerlov, C. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 2001, 107, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Fajas, L.; Egler, V.; Reiter, R.; Hansen, J.; Kristiansen, K.; Debril, M.B.; Miard, S.; Auwerx, J. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev. Cell 2002, 3, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Lyle, R.E.; McGehee, R.E. Regulation and expression of retinoblastoma proteins p107 and p130 during 3T3-L1 adipocyte differentiation. J. Biol. Chem. 1997, 272, 10117–10124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.L.; Riley, D.J.; Chen, Y.; Lee, W.H. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996, 10, 2794–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Classon, M.; Kennedy, B.K.; Mulloy, R.; Harlow, E. Opposing roles of pRB and p107 in adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2000, 97, 10826–10831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. p53 Functions in Adipose Tissue Metabolism and Homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallenborg, P.; Fjære, E.; Liaset, B.; Petersen, R.K.; Murano, I.; Sonne, S.B.; Falkerslev, M.; Winther, S.; Jensen, B.A.H.; Ma, T.; et al. p53 regulates expression of uncoupling protein 1 through binding and repression of PPARγ coactivator-1α. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E116–E128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molchadsky, A.; Ezra, O.; Amendola, P.G.; Krantz, D.; Kogan-Sakin, I.; Buganim, Y.; Rivlin, N.; Goldfinger, N.; Folgiero, V.; Falcioni, R.; et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ. 2013, 20, 774–783. [Google Scholar] [CrossRef]
- Molchadsky, A.; Shats, I.; Goldfinger, N.; Pevsner-Fischer, M.; Olson, M.; Rinon, A.; Tzahor, E.; Lozano, G.; Zipori, D.; Sarig, R.; et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE 2008, 3, e3707. [Google Scholar] [CrossRef] [Green Version]
- Okita, N.; Ishikawa, N.; Mizunoe, Y.; Oku, M.; Nagai, W.; Suzuki, Y.; Matsushima, S.; Mikami, K.; Okado, H.; Sasaki, T.; et al. Inhibitory effect of p53 on mitochondrial content and function during adipogenesis. Biochem. Biophys. Res. Commun. 2014, 446, 91–97. [Google Scholar] [CrossRef]
- Gustafson, B.; Nerstedt, A.; Smith, U. Reduced subcutaneous adipogenesis in human hypertrophic obesity is linked to senescent precursor cells. Nat. Commun. 2019, 10, 2757. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Yoshida, Y.; Katsuno, T.; Tateno, K.; Okada, S.; Moriya, J.; Yokoyama, M.; Nojima, A.; Ito, T.; Zechner, R.; et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012, 15, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Phelps, D.E.; Xiong, Y. Regulation of cyclin-dependent kinase 4 during adipogenesis involves switching of cyclin D subunits and concurrent binding of p18INK4c and p27Kip1. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1998, 9, 595–610. [Google Scholar]
- Lagarrigue, S.; Lopez-Mejia, I.C.; Denechaud, P.D.; Escoté, X.; Castillo-Armengol, J.; Jimenez, V.; Chavey, C.; Giralt, A.; Lai, Q.; Zhang, L.; et al. CDK4 is an essential insulin effector in adipocytes. J. Clin. Investig. 2016, 126, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Cudejko, C.; Wouters, K.; Fuentes, L.; Hannou, S.A.; Paquet, C.; Bantubungi, K.; Bouchaert, E.; Vanhoutte, J.; Fleury, S.; Remy, P.; et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood 2011, 118, 2556–2566. [Google Scholar] [CrossRef]
- Fuentes, L.; Wouters, K.; Hannou, S.A.; Cudejko, C.; Rigamonti, E.; Mayi, T.H.; Derudas, B.; Pattou, F.; Chinetti-Gbaguidi, G.; Staels, B.; et al. Downregulation of the tumour suppressor p16INK4A contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia 2011, 54, 3150–3156. [Google Scholar] [CrossRef]
- Li, L.; Ng, D.S.W.; Mah, W.C.; Almeida, F.F.; Rahmat, S.A.; Rao, V.K.; Leow, S.C.; Laudisi, F.; Peh, M.T.; Goh, A.M.; et al. A unique role for p53 in the regulation of M2 macrophage polarization. Cell Death Differ. 2015, 22, 1081–1093. [Google Scholar] [CrossRef]
- Blanchet, E.; Annicotte, J.S.; Lagarrigue, S.; Aguilar, V.; Clapé, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef] [Green Version]
- Rabhi, N.; Hannou, S.A.; Gromada, X.; Salas, E.; Yao, X.; Oger, F.; Carney, C.; Lopez-Mejia, I.C.; Durand, E.; Rabearivelo, I.; et al. Cdkn2a deficiency promotes adipose tissue browning. Mol. Metab. 2018, 8, 65–76. [Google Scholar] [CrossRef]
- Dali-Youcef, N.; Mataki, C.; Coste, A.; Messaddeq, N.; Giroud, S.; Blanc, S.; Koehl, C.; Champy, M.F.; Chambon, P.; Fajas, L.; et al. Adipose tissue-specific inactivation of the retinoblastoma protein protects against diabesity because of increased energy expenditure. Proc. Natl. Acad. Sci. USA 2007, 104, 10703–10708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanssen, M.J.W.; Hoeks, J.; Brans, B.; van der Lans, A.A.J.J.; Schaart, G.; van den Driessche, J.J.; Jörgensen, J.A.; Boekschoten, M.V.; Hesselink, M.K.C.; Havekes, B.; et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat. Med. 2015, 21, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci. Lond. Engl. 2020, 134, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Vergoni, B.; Cornejo, P.J.; Gilleron, J.; Djedaini, M.; Ceppo, F.; Jacquel, A.; Bouget, G.; Ginet, C.; Gonzalez, T.; Maillet, J.; et al. DNA Damage and the Activation of the p53 Pathway Mediate Alterations in Metabolic and Secretory Functions of Adipocytes. Diabetes 2016, 65, 3062–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawik, M.; Vidal-Puig, A.J. Adipose tissue expandability and the metabolic syndrome. Genes Nutr. 2007, 2, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I.; Rossi, D.J. Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease. Cell Stem Cell 2015, 16, 613. [Google Scholar] [CrossRef] [Green Version]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Rane, S.G.; Dubus, P.; Mettus, R.V.; Galbreath, E.J.; Boden, G.; Reddy, E.P.; Barbacid, M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef]
- Mettus, R.V.; Rane, S.G. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene 2003, 22, 8413–8421. [Google Scholar] [CrossRef] [Green Version]
- Martín, J.; Hunt, S.L.; Dubus, P.; Sotillo, R.; Néhmé-Pélluard, F.; Magnuson, M.A.; Parlow, A.F.; Malumbres, M.; Ortega, S.; Barbacid, M. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 2003, 22, 5261–5269. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.B.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [Green Version]
- Haim, Y.; Blüher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Kim, J.A.; Hong, S.; Lee, B.; Hong, J.W.; Kwak, J.Y.; Cho, S.; Kim, C.C. The inhibition of T-cells proliferation by mouse mesenchymal stem cells through the induction of p16INK4A-cyclin D1/cdk4 and p21waf1, p27kip1-cyclin E/cdk2 pathways. Cell. Immunol. 2007, 245, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Yoshida, Y.; Moriya, J.; Nojima, A.; Uemura, A.; Kobayashi, Y.; Minamino, T. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab. 2013, 18, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Liu, Y.; Sun, C.; Yin, H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 844–856. [Google Scholar] [CrossRef]
- Berry, D.C.; Jiang, Y.; Arpke, R.W.; Close, E.L.; Uchida, A.; Reading, D.; Berglund, E.D.; Kyba, M.; Graff, J.M. Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell Metab. 2017, 25, 166–181. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell. Physiol. 2018, 234, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimè, A.; Grenier, G.; Huh, M.S.; Gillespie, M.A.; Bevilacqua, L.; Harper, M.E.; Rudnicki, M.A. Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1alpha. Cell Metab. 2005, 2, 283–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, Y.; Asano, A.; Cañas, X.; Nikami, H.; Aizawa, S.; Saito, M. Immortal brown adipocytes from p53-knockout mice: Differentiation and expression of uncoupling proteins. Biochem. Biophys. Res. Commun. 1999, 255, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kim, T.H.; Franklin, D.A.; Zhang, Y. Protection against High-Fat-Diet-Induced Obesity in MDM2C305F Mice Due to Reduced p53 Activity and Enhanced Energy Expenditure. Cell Rep. 2017, 18, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- González-Navarro, H.; Vinué, Á.; Sanz, M.J.; Delgado, M.; Pozo, M.A.; Serrano, M.; Burks, D.J.; Andrés, V. Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with aging. Aging Cell 2013, 12, 102–111. [Google Scholar] [CrossRef]
- Pal, A.; Potjer, T.P.; Thomsen, S.K.; Ng, H.J.; Barrett, A.; Scharfmann, R.; James, T.J.; Bishop, D.T.; Karpe, F.; Godsland, I.F.; et al. Loss-of-Function Mutations in the Cell-Cycle Control Gene CDKN2A Impact on Glucose Homeostasis in Humans. Diabetes 2016, 65, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Aguayo-Mazzucato, C.; van Haaren, M.; Mruk, M.; Lee, T.B.; Crawford, C.; Hollister-Lock, J.; Sullivan, B.A.; Johnson, J.W.; Ebrahimi, A.; Dreyfuss, J.M.; et al. β Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. 2017, 25, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Sharma, R.B.; Ly, S.; Stamateris, R.E.; Jesdale, W.M.; Alonso, L.C. CDKN2A/B T2D Genome-Wide Association Study Risk SNPs Impact Locus Gene Expression and Proliferation in Human Islets. Diabetes 2018, 67, 872–884. [Google Scholar] [CrossRef] [Green Version]
- Bantubungi, K.; Hannou, S.A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1α pathway. Diabetes 2014, 63, 3199–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Mohlke, K.L.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS ONE 2009, 4, e5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Navarro, H.; Abu Nabah, Y.N.; Vinué, A.; Andrés-Manzano, M.J.; Collado, M.; Serrano, M.; Andrés, V. p19(ARF) deficiency reduces macrophage and vascular smooth muscle cell apoptosis and aggravates atherosclerosis. J. Am. Coll. Cardiol. 2010, 55, 2258–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.L.; Murphy, A.J.; Sayers, S.; Li, R.; Yvan-Charvet, L.; Davis, J.Z.; Krishnamurthy, J.; Liu, Y.; Puig, O.; Sharpless, N.E.; et al. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2483–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, e2002940. [Google Scholar] [CrossRef] [Green Version]
- Le, O.; Palacio, L.; Bernier, G.; Batinic-Haberle, I.; Hickson, G.; Beauséjour, C. INK4a/ARF Expression Impairs Neurogenesis in the Brain of Irradiated Mice. Stem Cell Rep. 2018, 10, 1721–1733. [Google Scholar] [CrossRef] [Green Version]
- Price, J.D.; Park, K.Y.; Chen, J.; Salinas, R.D.; Cho, M.J.; Kriegstein, A.R.; Lim, D.A. The Ink4a/Arf locus is a barrier to direct neuronal transdifferentiation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 12560–12567. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.A.; Gomis-Coloma, C.; Morenilla-Palao, C.; Peiro, G.; Serra, E.; Serrano, M.; Cabedo, H. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain J. Neurol. 2013, 136, 2262–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VinuÉ, Á.; MartÍnez-HervÁs, S.; Herrero-Cervera, A.; SÁnchez-GarcÍa, V.; AndrÉs-Blasco, I.; Piqueras, L.; Sanz, M.J.; Real, J.T.; Ascaso, J.F.; Burks, D.J.; et al. Changes in CDKN2A/2B expression associate with T-cell phenotype modulation in atherosclerosis and type 2 diabetes mellitus. Transl. Res. J. Lab. Clin. Med. 2019, 203, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hervás, S.; Sánchez-García, V.; Herrero-Cervera, A.; Vinué, Á.; Real, J.T.; Ascaso, J.F.; Burks, D.J.; González-Navarro, H. Type 1 diabetic mellitus patients with increased atherosclerosis risk display decreased CDKN2A/2B/2BAS gene expression in leukocytes. J. Transl. Med. 2019, 17, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Nakagawa, H.; Tajima, A.; Yoshida, K.; Inoue, I. ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol. Rep. 2010, 24, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.H.; Tang, J.M.; Li, J.; Li, X.W. Upregulation of SOX2-activated lncRNA ANRIL promotes nasopharyngeal carcinoma cell growth. Sci. Rep. 2018, 8, 3333. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Hsieh, C.H.; Alonso, L.C. ANRIL: A lncRNA at the CDKN2A/B Locus With Roles in Cancer and Metabolic Disease. Front. Endocrinol. 2018, 9, 405. [Google Scholar] [CrossRef] [Green Version]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.T.; Xu, J.H.; Zheng, Z.R.; Zhao, Q.Q.; Zeng, X.S.; Cheng, S.X.; Liang, Y.H.; Hu, Q.F. Long non-coding RNA ANRIL promotes carcinogenesis via sponging miR-199a in triple-negative breast cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 96, 14–21. [Google Scholar] [CrossRef]
- Shi, C.; Huang, F.; Gu, X.; Zhang, M.; Wen, J.; Wang, X.; You, L.; Cui, X.; Ji, C.; Guo, X. Adipogenic miRNA and meta-signature miRNAs involved in human adipocyte differentiation and obesity. Oncotarget 2016, 7, 40830–40845. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.D.; Dewal, R.S.; Stanford, K.I. The beneficial effects of brown adipose tissue transplantation. Mol. Asp. Med. 2019, 68, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Petkova, A.P.; Mottillo, E.P.; Granneman, J.G. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012, 15, 480–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahoul, Y.; Oger, F.; Montaigne, J.; Froguel, P.; Breton, C.; Annicotte, J.-S. Emerging Roles for the INK4a/ARF (CDKN2A) Locus in Adipose Tissue: Implications for Obesity and Type 2 Diabetes. Biomolecules 2020, 10, 1350. https://doi.org/10.3390/biom10091350
Kahoul Y, Oger F, Montaigne J, Froguel P, Breton C, Annicotte J-S. Emerging Roles for the INK4a/ARF (CDKN2A) Locus in Adipose Tissue: Implications for Obesity and Type 2 Diabetes. Biomolecules. 2020; 10(9):1350. https://doi.org/10.3390/biom10091350
Chicago/Turabian StyleKahoul, Yasmina, Frédérik Oger, Jessica Montaigne, Philippe Froguel, Christophe Breton, and Jean-Sébastien Annicotte. 2020. "Emerging Roles for the INK4a/ARF (CDKN2A) Locus in Adipose Tissue: Implications for Obesity and Type 2 Diabetes" Biomolecules 10, no. 9: 1350. https://doi.org/10.3390/biom10091350