A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin

 ,
,  , ,
, ,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Protein Expression and Purification
2.3. Design and Synthesis of the Peptides
2.4. Fluorescence
2.4.1. Steady-State Fluorescence
2.4.2. Thermal Denaturations
2.5. CD
2.5.1. Far-Ultraviolet (UV) Spectra
2.5.2. Thermal Denaturations
2.6. ITC
2.7. NMR
2.7.1. 1D-1H-NMR (One-Dimensional Proton NMR) Spectra
2.7.2. Translational NMR Diffusion Ordered Spectroscopy (DOSY)
2.7.3. 2D-1H-NMR Spectra
2.8. Molecular Docking
3. Results
3.1. The Isolated wt NLS-NUPR1 and Its Mutants Were Monomeric and Disordered in Aqueous Solution
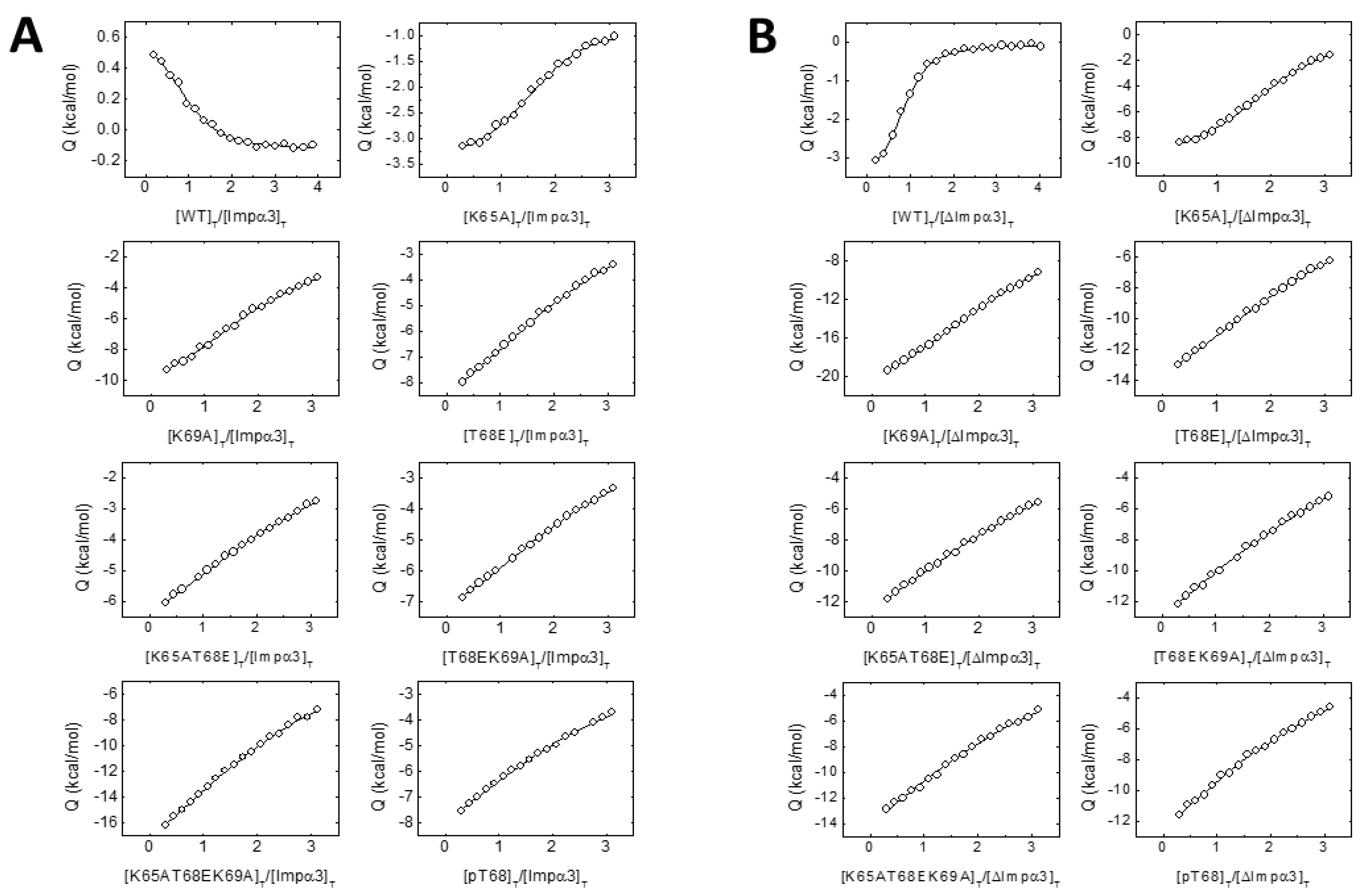
3.2. The NLS-NUPR1 Peptides Bound to Both Impα3 and ΔImpα3
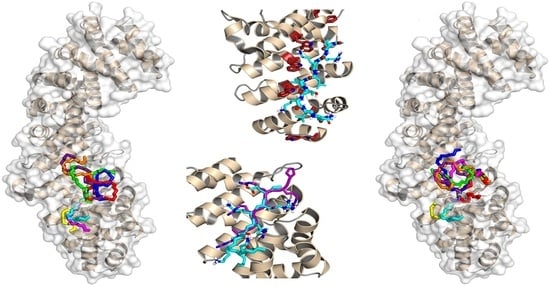
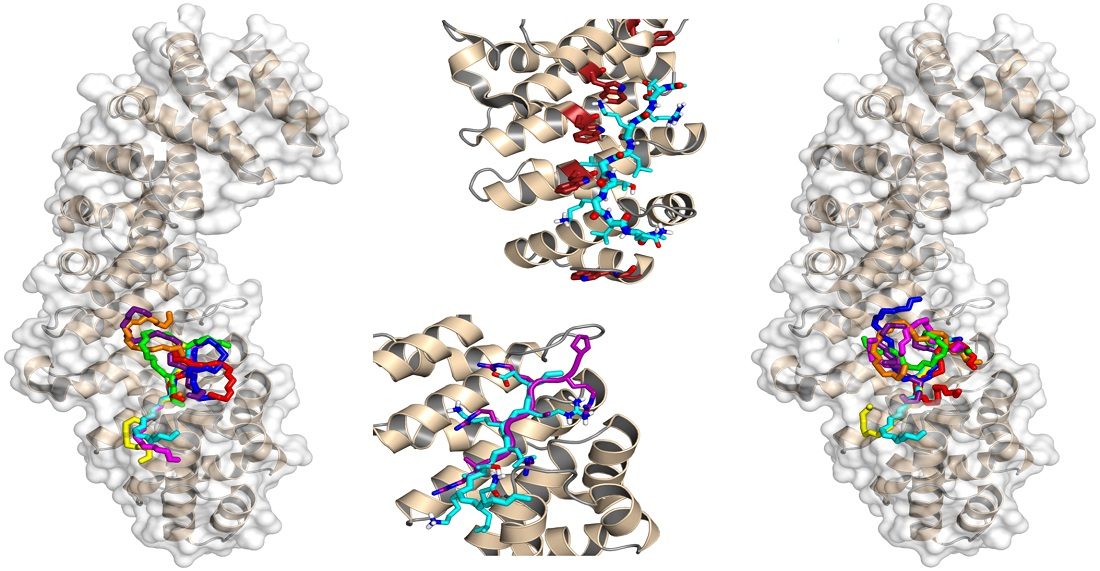
3.3. Binding Regions in the Docking of NUPR1 Peptides to Importins
4. Discussion
4.1. Molecular Mechanisms for Impα3 Recognition of NUPR1: The Influence of Lys65 and Lys69
4.2. Molecular Mechanisms for Impα3 Recognition of NUPR1: The Influence of Thr68 and Its Phosphorylation-Triggered Conformational Switch
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Boil. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Bednenko, J.; Cingolari, G.; Gerace, L. Nucleo-cytoplasmic transport navigating the channel. Traffic 2003, 4, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, G.; Bednenko, J.; Gillespie, M.T.; Gerace, L. Molecular basis for the recognition of a non-classical nuclear localization signal by importin beta. Mol. Cell 2002, 10, 1345–1353. [Google Scholar] [CrossRef]
- Goldfarb, D.S.; Corbett, A.H.; Mason, D.A.; Harreman, M.T.; Adam, S.A. Importin α: A multipurpose nuclear-transport receptor. Trends Cell Boil. 2004, 14, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Pumroy, R.A.; Cingolani, G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 2015, 466, 13–28. [Google Scholar] [CrossRef] [Green Version]
- Mason, D.A.; Stage, D.E.; Goldfarb, D. Evolution of the metazoan-specific importin α gene family. J. Mol. Evol. 2009, 68, 351–365. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Loveland, K.L.; Yoneda, Y. Nuclear importin α and its physiological importance. Commun. Integr. Boil. 2012, 5, 220–222. [Google Scholar] [CrossRef]
- Smith, K.M.; Tsimbalyuk, S.; Edwards, M.R.; Cross, E.M.; Batra, J.; Da Costa, T.P.S.; Aragão, D.; Basler, C.; Forwood, J. Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat. Commun. 2018, 9, 3703. [Google Scholar] [CrossRef]
- Kobe, B. Autoinhibition by an internal nuclear localization signal revealed by the crystal structure of mammalian importin α. Nat. Struct. Biol. 1999, 6, 388–397. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Boil. 2018, 430, 2309–2320. [Google Scholar] [CrossRef]
- Xie, H.M.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic†, Z. Functional Anthology of Intrinsic Disorder. 1. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res. 2007, 6, 1882–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M.; Van Der Lee, R.; De Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Boil. 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight Regulation of Unstructured Proteins: From Transcript Synthesis to Protein Degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [Green Version]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-translational Modifications. J. Boil. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [Green Version]
- Launay, H.; Receveur-Bréchot, V.; Carrière, F.; Gontero, B. Orchestration of algal metabolism by protein disorder. Arch. Biochem. Biophys. 2019, 672, 108070. [Google Scholar] [CrossRef]
- Mallo, G.V.; Fiedler, F.; Calvo, E.L.; Ortiz, E.M.; Vasseur, S.; Keim, V.; Morisset, J.; Iovanna, J.L. Cloning and Expression of the Rat p8 cDNA, a New Gene Activated in Pancreas during the Acute Phase of Pancreatitis, Pancreatic Development, and Regeneration, and Which Promotes Cellular Growth. J. Boil. Chem. 1997, 272, 32360–32369. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, U.R.; Samant, R.S.; Fodstad, O.; Shevde, L.A. Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev. 2009, 28, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Iovanna, J.L. Stress-inducible Protein p8 Is Involved in Several Physiological and Pathological Processes. J. Boil. Chem. 2009, 285, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Cano, C.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-knife of cancer. J. Cell. Physiol. 2010, 226, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Malicet, C.; Giroux, V.; Vasseur, S.; Dagorn, J.C.; Neira, J.L.; Iovanna, J.L. Regulation of apoptosis by the p8/prothymosin alpha complex. Proc. Natl. Acad. Sci. USA 2006, 103, 2671–2676. [Google Scholar] [CrossRef] [Green Version]
- Encinar, J.A.; Mallo, G.V.; Mizyrycki, C.; Giono, L.E.; González-Ros, J.M.; Rico, M.; Cánepa, E.T.; Moreno, S.; Neira, J.L.; Iovanna, J.L. Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J. Boil. Chem. 2000, 276, 2742–2751. [Google Scholar] [CrossRef] [Green Version]
- Aguado-Llera, D.; Hamidi, T.; Doménech, R.; Pantoja-Uceda, D.; Gironella, M.; Santoro, J.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J.L. Deciphering the binding between Nupr1 and MSL1 and Their DNA-Repairing Activity. PLoS ONE 2013, 8, e78101. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an intrinsically disordered protein involved in pancreatic adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Rizzuti, B.; Pey, A.L.; Soubeyran, P.; Vidal, M.; Urrutia, R.; Iovanna, J.L.; Neira, J.L. Intrinsically disordered chromatin protein NUPR1 binds to the C-terminal region of Polycomb RING1B. Proc. Natl. Acad. Sci. USA 2017, 114, E6332–E6341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valacco, M.P.; Varone, C.L.; Malicet, C.; Cánepa, E.T.; Iovanna, J.L.; Moreno, S. Cell growth-dependent subcellular localization of p8. J. Cell. Biochem. 2006, 97, 1066–1079. [Google Scholar] [CrossRef]
- Jäkel, S.; Mingot, J.-M.; Schwarzmaier, P.; Hartmann, E.; Görlich, D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 2002, 21, 377–386. [Google Scholar] [CrossRef]
- Marvaldi, L.; Panayotis, N.; Alber, S.; Dagan, S.Y.; Okladnikov, N.; Koppel, I.; Di Pizio, A.; Song, D.-A.; Tzur, Y.; Terenzio, M.; et al. Importin α3 regulates chronic pain pathways in peripheral sensory neurons. Science 2020, 369, 842–846. [Google Scholar] [CrossRef]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Gräslund, A. Translational diffusion measured by PFG-NMR on full length and fragments of the Alzheimer Aβ(1-40) peptide. Determination of hydrodynamic radii of random coil peptides of varying length. Magn. Reson. Chem. 2002, 40, S89–S97. [Google Scholar] [CrossRef]
- Neira, J.L.; Hornos, F.; Bacarizo, J.; Camara-Artigas, A.; Gómez, J. The monomeric species of the regulatory domain of Tyrosine Hydroxylase has a low conformational stability. Biochemistry 2016, 55, 3418–3431. [Google Scholar] [CrossRef]
- Benjwal, S.; Verma, S.; Röhm, K.; Gursky, O. Monitoring protein aggregation during thermal unfolding in circular dichroism experiments. Protein Sci. 2006, 15, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castaño, P.; Xia, Y.; Lan, W.; Zhou, Z.; Huang, C.; Peng, L.; Soubeyran, P.; Velázquez-Campoy, A.; Abian, O.; Rizzuti, B.; et al. Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J. Clin. Investig. 2019, 129, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, J.; Fairbrother, W.J.; Palmer, A.G.; Skelton, N.J. Protein NMR Spectroscopy: Principles and Practice; Academic Press: New York, NY, USA, 1996. [Google Scholar]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Marion, D.; Wüthrich, K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 1983, 113, 967–974. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.; Wüthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Cavanagh, J.; Rance, M. Suppression of cross-relaxation effects in TOCSY spectra via a modified DIPSI-2 mixing sequence. J. Magn. Reson. 1992, 96, 670–678. [Google Scholar] [CrossRef]
- Piotto, M.; Saudek, V.; Sklenář, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 1992, 2, 661–665. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley and Sons: New York, NY, USA, 1986. [Google Scholar]
- Kjaergaard, M.; Brander, S.; Poulsen, F.M. Random coil chemical shift for intrinsically disordered proteins: Effects of temperature and pH. J. Biomol. NMR 2011, 49, 139–149. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Poulsen, F.M. Sequence correction of random coil chemical shifts: Correlation between neighbor correction factors and changes in the Ramachandran distribution. J. Biomol. NMR 2011, 50, 157–165. [Google Scholar] [CrossRef]
- Bienkiewicz, E.A.; Lumb, K.J. Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 1999, 15, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Hendus-Altenburger, R.; Fernandes, C.B.; Bugge, K.; Kunze, M.B.A.; Boomsma, W.; Kragelund, B.B. Random coil chemical shifts for serine, threonine and tyrosine phosphorylation over a broad pH range. J. Biomol. NMR 2019, 73, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, R.; Matsuura, Y. Crystal structure of importin--α bound to the nuclear localization signal of Epstein--Barr virus EBNA--LP protein. Protein Sci. 2017, 26, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by molecular docking of homoisoflavones from Leopoldia comosa as ligands of estrogen receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [Green Version]
- Grimsley, G.R.; Huyghues-Despointes, B.M.; Pace, C.N.; Scholtz, J.M. Measuring the Conformational Stability of a Protein by NMR. Cold Spring Harb. Protoc. 2006, 2006, 253–259. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Swayden, M.; Xia, Y.; Zhou, Z.; Audebert, S.; Camoin, L.; Huang, C.; Peng, L.; Jiménez-Alesanco, A.; et al. ZZW-115-dependent inhibition of NUPR1 nuclear translocation sensitizes cancer cells to genotoxic agents. JCI Insight 2020, 138117. [Google Scholar] [CrossRef]
- Neira, J.L.; López, M.B.; Sevilla, P.; Rizzuti, B.; Camara-Artigas, A.; Vidal, M.; Iovanna, J.L. The chromatin nuclear protein NUPR1L is intrinsically disordered and binds to the same proteins as its paralogue. Biochem. J. 2018, 475, 2271–2291. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Abian, O.; Velázquez-Campoy, A.; Iovanna, J.L.; Neira, J.L. Amphipathic helical peptides hamper protein-protein interactions of the intrinsically disordered chromatin nuclear protein 1 (NUPR1). Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1283–1295. [Google Scholar] [CrossRef] [Green Version]
- Miyatake, H.; Sanjoh, A.; Unzai, S.; Matsuda, G.; Tatsumi, Y.; Miyamoto, Y.; Dohmae, N.; Aida, Y. Crystal structure of human Importin-α1 (Rch1), revealing a potential autoinhibition mode involving homodimerization. PLoS ONE 2015, 10, e0115995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankhala, R.S.; Lokareddy, R.K.; Begum, S.; Pumroy, R.A.; Gillilan, R.E.; Cingolani, G. Three-dimensional context rather than NLS amino acid sequence determines importin α subtype specificity for RCC1. Nat. Commun. 2017, 8, 979. [Google Scholar] [CrossRef] [PubMed]
- Pumroy, R.A.; Ke, S.; Hart, D.J.; Zacharie, U.; Cingolani, G. Molecular determinants for nuclear import if influenza A PB2 by importin alpha isoforms 3 and 7. Structure 2015, 23, 374–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junod, S.L.; Kelich, J.M.; Ma, J.; Yang, W. Nucleocytoplasmic transport of intrinsically disordered proteins studied by high--speed super--resolution microscopy. Protein Sci. 2020, 29, 1459–1472. [Google Scholar] [CrossRef]
- Neira, J.L.; Correa, J.; Rizzuti, B.; Santofimia-Castaño, P.; Abián, O.; Velázquez-Campoy, A.; Fernandez-Megia, E.; Iovanna, J.L. Dendrimers as competitors of protein–protein interactions of the intrinsically disordered nuclear chromatin protein NUPR1. Biomacromolecules 2019, 20, 2567–2576. [Google Scholar] [CrossRef]
- Yadahalli, S.; Neira, J.L.; Johnson, C.M.; Tan, Y.S.; Rowling, P.J.E.; Chattopadhyay, A.; Verma, C.; Itzhaki, L.S. Kinetic and thermodynamic effects of phosphorylation on p53 binding to MDM2. Sci. Rep. 2019, 9, 693. [Google Scholar] [CrossRef]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.W.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2014, 519, 106–109. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Landrieu, I.; Byrne, C.; Kukić, P.; Amniai, L.; Cantrelle, F.-X.; Wieruszeski, J.-M.; Mancera, R.L.; Jacquot, Y.; Lippens, G. A Phosphorylation-induced turn defines the Alzheimer’s disease AT8 antibody epitope on the Tau protein. Angew. Chem. Int. Ed. 2015, 54, 6819–6823. [Google Scholar] [CrossRef]
- Beck, D.A.C.; Alonso, D.O.V.; Inoyama, D.; Daggett, V. The intrinsic conformational propensities of the 20 naturally occurring amino acids and reflection of these propensities in proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12259–12264. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, V.; Serrano, L. Intrinsic secondary structure propensities of the amino acids, using statistical φ-ψ matrices: Comparison with experimental scales. Proteins: Struct. Funct. Bioinform. 1994, 20, 301–311. [Google Scholar] [CrossRef]
- Gibbs, E.B.; Lu, F.; Portz, B.; Fisher, M.J.; Medellin, B.P.; Laremore, T.N.; Zhang, Y.S.; Gimour, D.S.; Showalter, S.A. Phosphorylation induces sequence-specific conformational switches in the RNA polymerase II C-terminal domain. Nat. Commun. 2017, 8, 15233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Gapsys, V.; Kim, H.-Y.; Bessonov, S.; Hsiao, H.-H.; Möhlmann, S.; Klaukien, V.; Ficner, R.; Becker, S.; Urlaub, H.; et al. Phosphorylation drives a dynamic switch in Serine/Arginine-rich proteins. Structure 2013, 21, 2162–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, A.L.; Watson, M.; Wilkins, O.G.; Cato, L.; Travers, A.; Thomas, J.O.; Stott, K. Highly disordered histone H1−DNA model complexes and their condensates. Proc. Natl. Acad. Sci. USA 2018, 115, 11964–11969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banavali, N.K.; Roux, B. Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins: Struct. Funct. Bioinform. 2007, 67, 1096–1112. [Google Scholar] [CrossRef]
- Espinoza-Fonseca, L.M.; Kast, D.; Thomas, D.D. Molecular dynamics simulations reveal a disorder-to-order transition on phosphorylation of smooth muscle myosin. Biophys. J. 2007, 93, 2083–2090. [Google Scholar] [CrossRef] [Green Version]
- Hendus-Altenburger, R.; Lambrughi, M.; Terkelsen, T.; Pedersen, S.F.; Papaleo, E.; Lindorff-Larsen, K.; Kragelund, B.B. A phosphorylation-motif for tuneable helix stabilisation in intrinsically disordered proteins–Lessons from the sodium proton exchanger 1 (NHE1). Cell. Signal. 2017, 37, 40–51. [Google Scholar] [CrossRef]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- He, Y.; Chen, Y.; Mooney, S.M.; Rajagopalan, K.; Bhargava, A.; Sacho, E.; Weninger, K.; Bryan, P.N.; Kulkarni, P.; Orban, J. Phosphorylation-induced conformational ensemble switching in an intrinsically disordered cancer/testis antigen*. J. Boil. Chem. 2015, 290, 25090–25102. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.T.; Williams, M.M.; Klee, E.W.; Lomberk, G.L.; Urrutia, R.A. Modeling post--translational modifications and cancer--associated mutations that impact the heterochromatin protein 1α--importin α heterodimers. Proteins: Struct. Funct. Bioinform. 2019, 87, 904–916. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide a | D (cm2 s−1) × 106 (Rh, Å) b | Rh, Å c |
|---|---|---|
| YT54NRPSPGGHERKLVTKLQNSE (wt) | 1.85 ± 0.04 (11 ± 1) | 13 ± 3 |
| YTNRPSPGGHERALVTKLQNSE (K65A) | 1.94 ± 0.08 (11 ± 1) | 13 ± 3 |
| YTNRPSPGGHERKLVTALQNSE (K69A) | 1.79 ± 0.06 (12 ± 2) | 13 ± 3 |
| YTNRPSPGGHERKLVEKLQNSE (T68E) | 2.17 ± 0.06 (10 ± 1) | 13 ± 3 |
| YTNRPSPGGHERALVEKLQNSE (K65AT68E) | 1.76 ± 0.06 (12 ± 1) | 13 ± 3 |
| YTNRPSPGGHERKLVEALQNSE (T68EK69A) | 1.87 ± 0.08 (11 ± 1) | 13 ± 3 |
| YTNRPSPGGHERALVEALQNSE (K65AT68EK69A) | 2.4 ± 0.2 (9 ± 2) | 13 ± 3 |
| YTNRPSPGGHERKLVpTKLQNSE (pT68) | 1.89 ± 0.08 (11 ± 1) | 13 ± 3 |
| Impα3 | ΔImpα3 | |||||||
|---|---|---|---|---|---|---|---|---|
| Peptide | Kd (μM) | ΔH (kcal/mol) | −TΔS (kcal/mol) | n | Kd (μM) | ΔH (kcal/mol) | −TΔS (kcal/mol) | n |
| wt | 1.7 | 0.8 | −8.7 | 0.9 | 0.95 | −3.7 | −4.5 | 1.0 |
| K65A | 3.9 | −2.8 | −4.6 | 1.4 | 2.7 | −10.2 | 2.6 | 1.4 |
| K69A | 11 | −10.8 | 4.0 | 1.3 | 7.6 | −21.3 | 14.3 | 1.4 |
| T68E | 22 | −11.1 | 4.7 | (1) | 12 | −17.5 | 10.8 | (1) |
| K65AT68E | 21 | −7.8 | 1.4 | (1) | 14 | −17.9 | 11.3 | (1) |
| T68EK69A | 17 | −7.5 | 1.0 | (1) | 17 | −21.2 | 14.7 | (1) |
| K65AT68EK69A | 27 | −16.3 | 9.1 | (1) | 24 | −28.5 | 22.2 | (1) |
| pT68 | 27 | −14.8 | 3.6 | (1) | 29 | −28.2 | 22.0 | (1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neira, J.L.; Rizzuti, B.; Jiménez-Alesanco, A.; Palomino-Schätzlein, M.; Abián, O.; Velázquez-Campoy, A.; Iovanna, J.L. A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin. Biomolecules 2020, 10, 1313. https://doi.org/10.3390/biom10091313
Neira JL, Rizzuti B, Jiménez-Alesanco A, Palomino-Schätzlein M, Abián O, Velázquez-Campoy A, Iovanna JL. A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin. Biomolecules. 2020; 10(9):1313. https://doi.org/10.3390/biom10091313
Chicago/Turabian StyleNeira, José L., Bruno Rizzuti, Ana Jiménez-Alesanco, Martina Palomino-Schätzlein, Olga Abián, Adrián Velázquez-Campoy, and Juan L. Iovanna. 2020. "A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin" Biomolecules 10, no. 9: 1313. https://doi.org/10.3390/biom10091313