Lifestyle Characteristics and Gene Expression Analysis of Colletotrichum camelliae Isolated from Tea Plant [Camellia sinensis (L.) O. Kuntze] Based on Transcriptome

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Microscopy Observation
2.3. Pathogen Infection Assay
2.4. RNA Isolation and Sequencing
2.5. Transcriptome Data Analysis
2.6. qRT-PCR Verification Assay for Candidate Effector Proteins (CEPs)
2.7. Cloning and Subcellular Localization of Candidate Effectors
3. Results
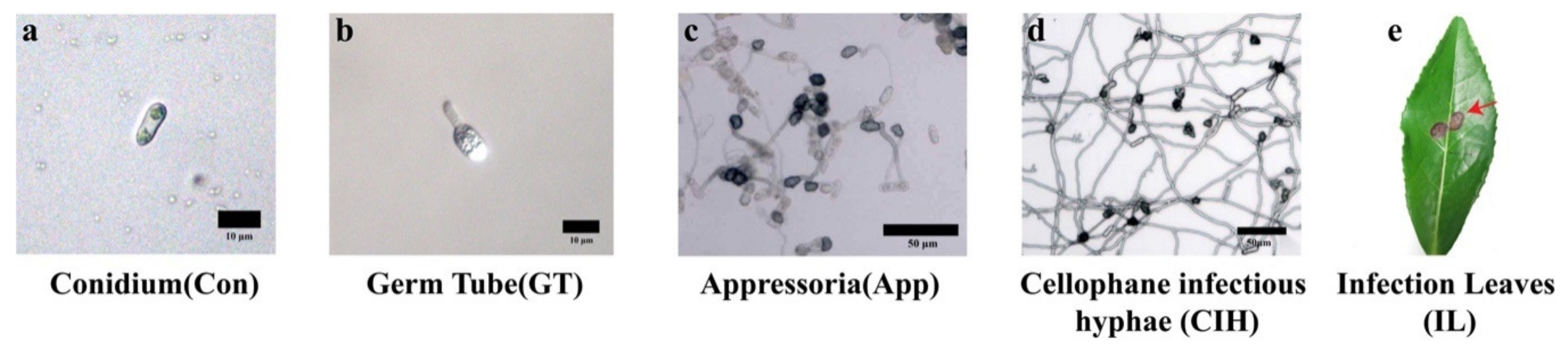
3.1. Different Developmental Stages of C. camelliae
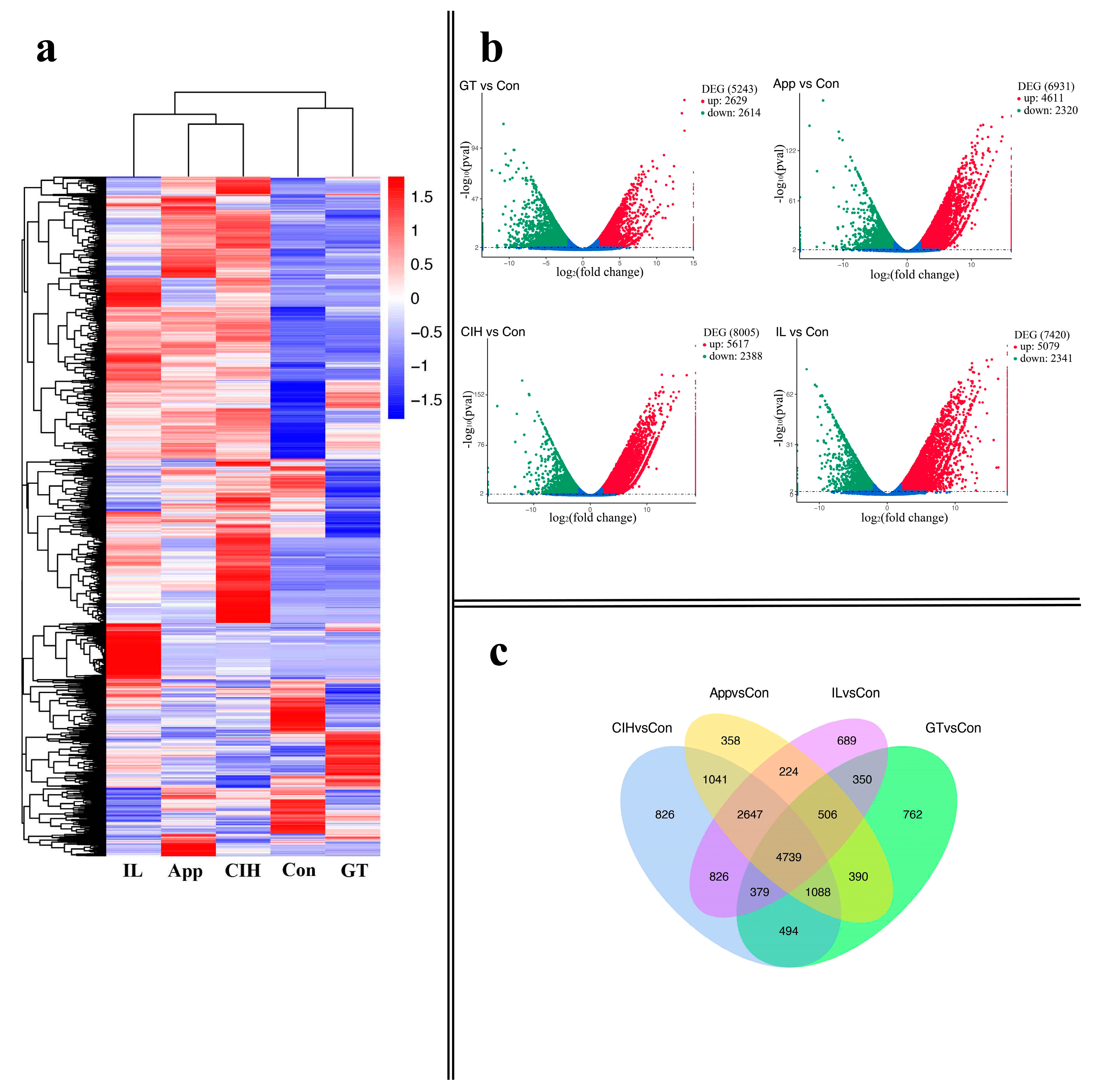
3.2. Transcriptome Assembly and Identification of Differentially Expressed Genes (DEGs) among Different Growth Stages
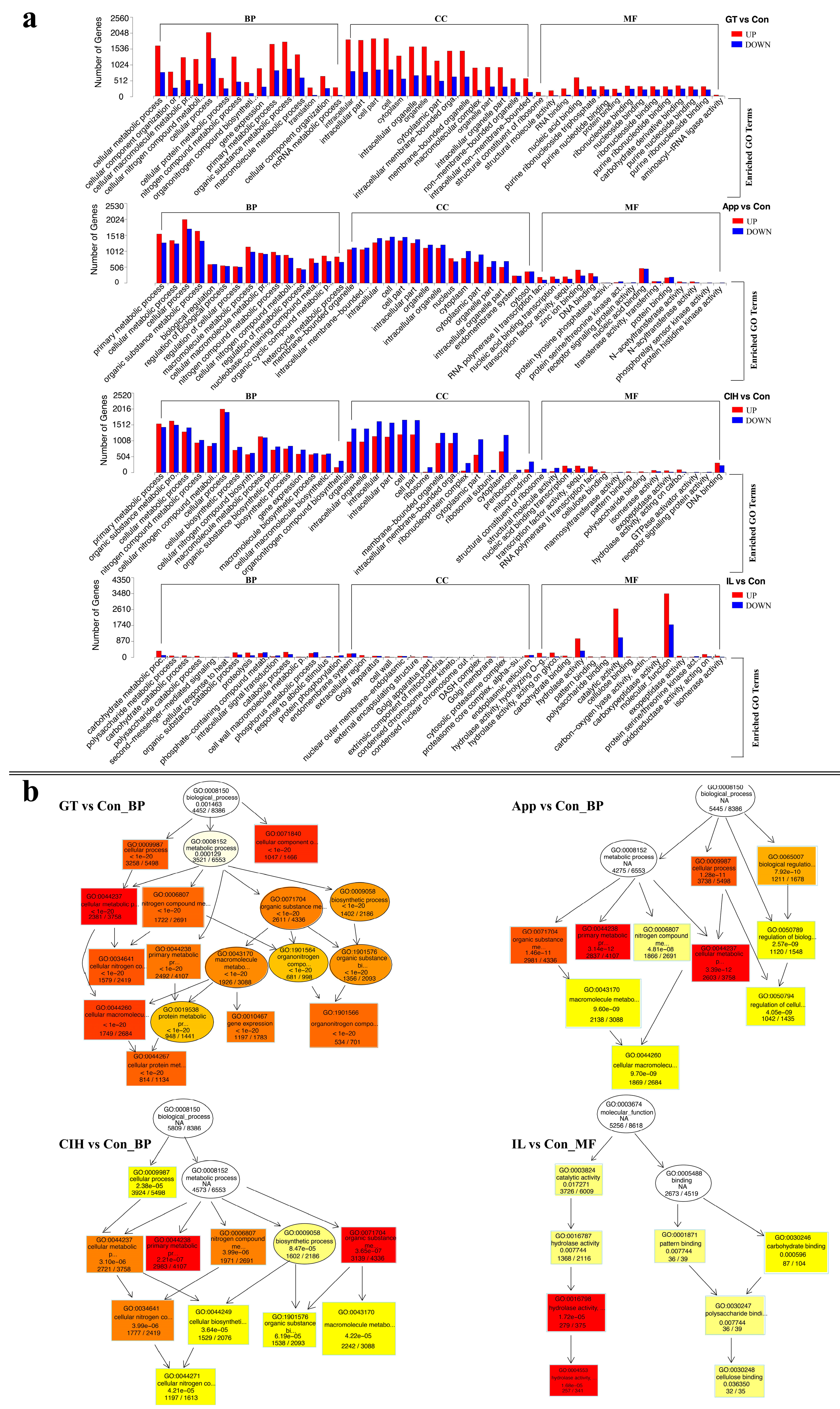
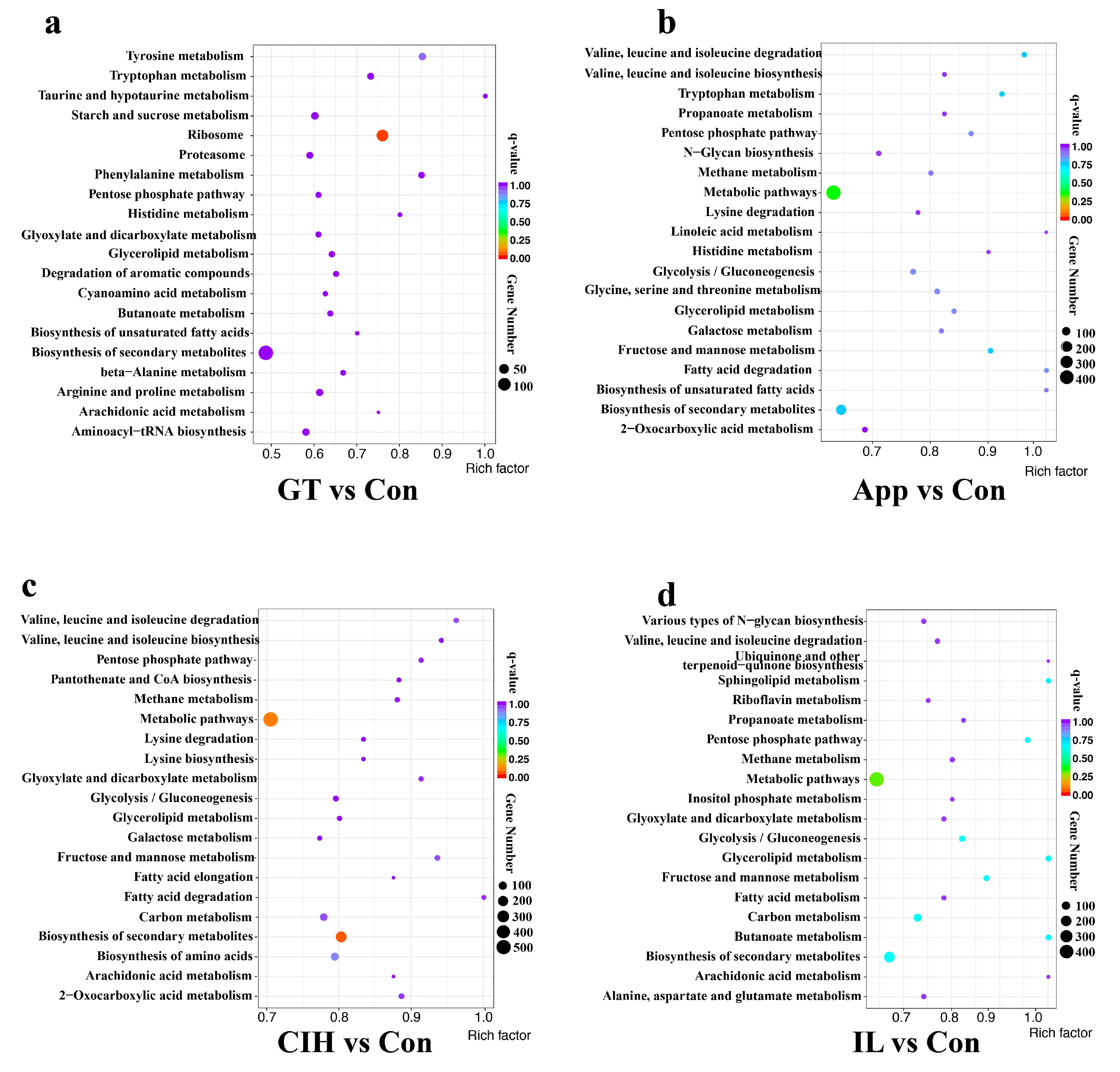
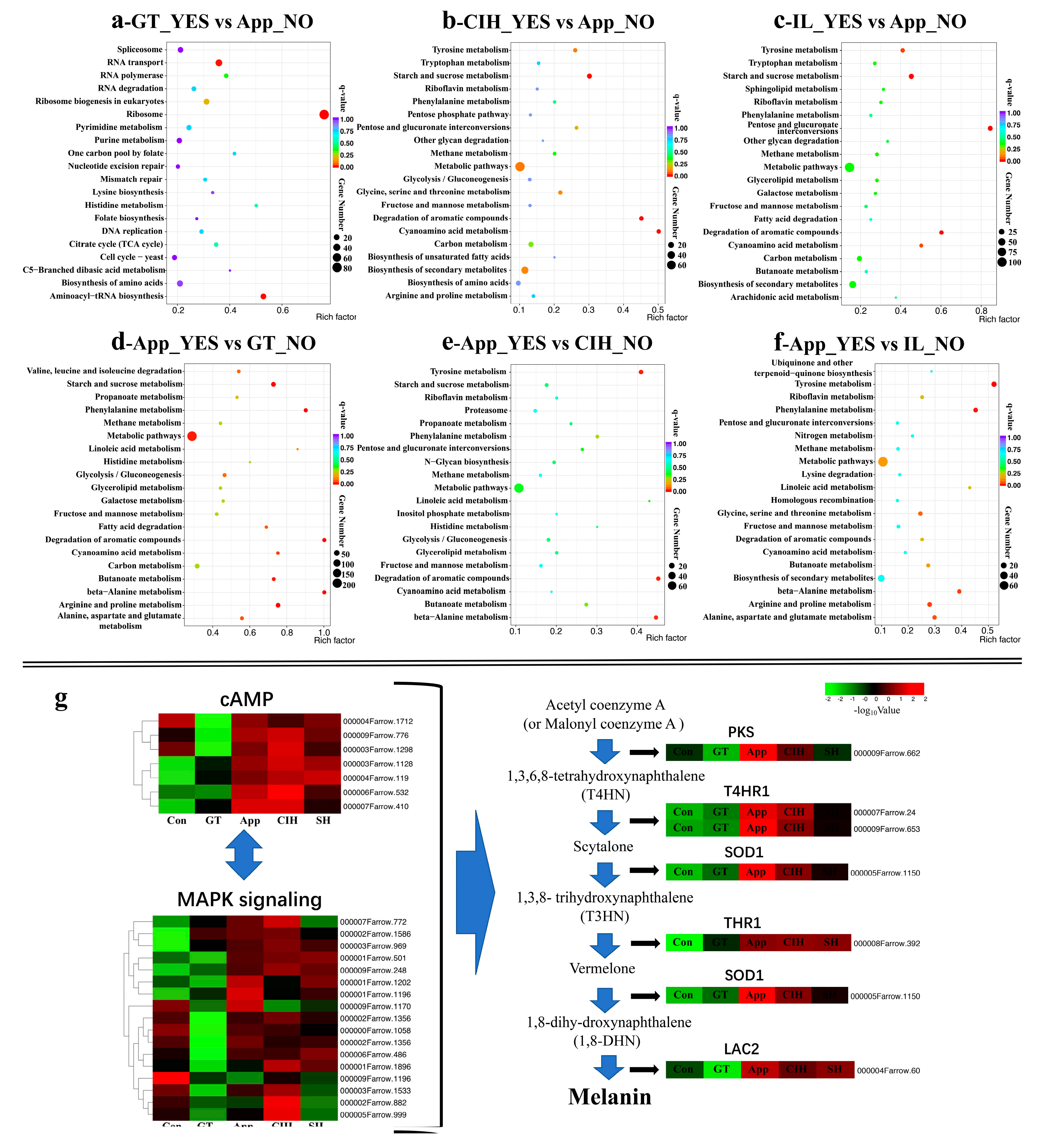
3.3. Functional Enrichment Reveals the Importance of Cellular Processes and Metabolic Pathways
3.4. Infection Strategy during Different Infection Stages
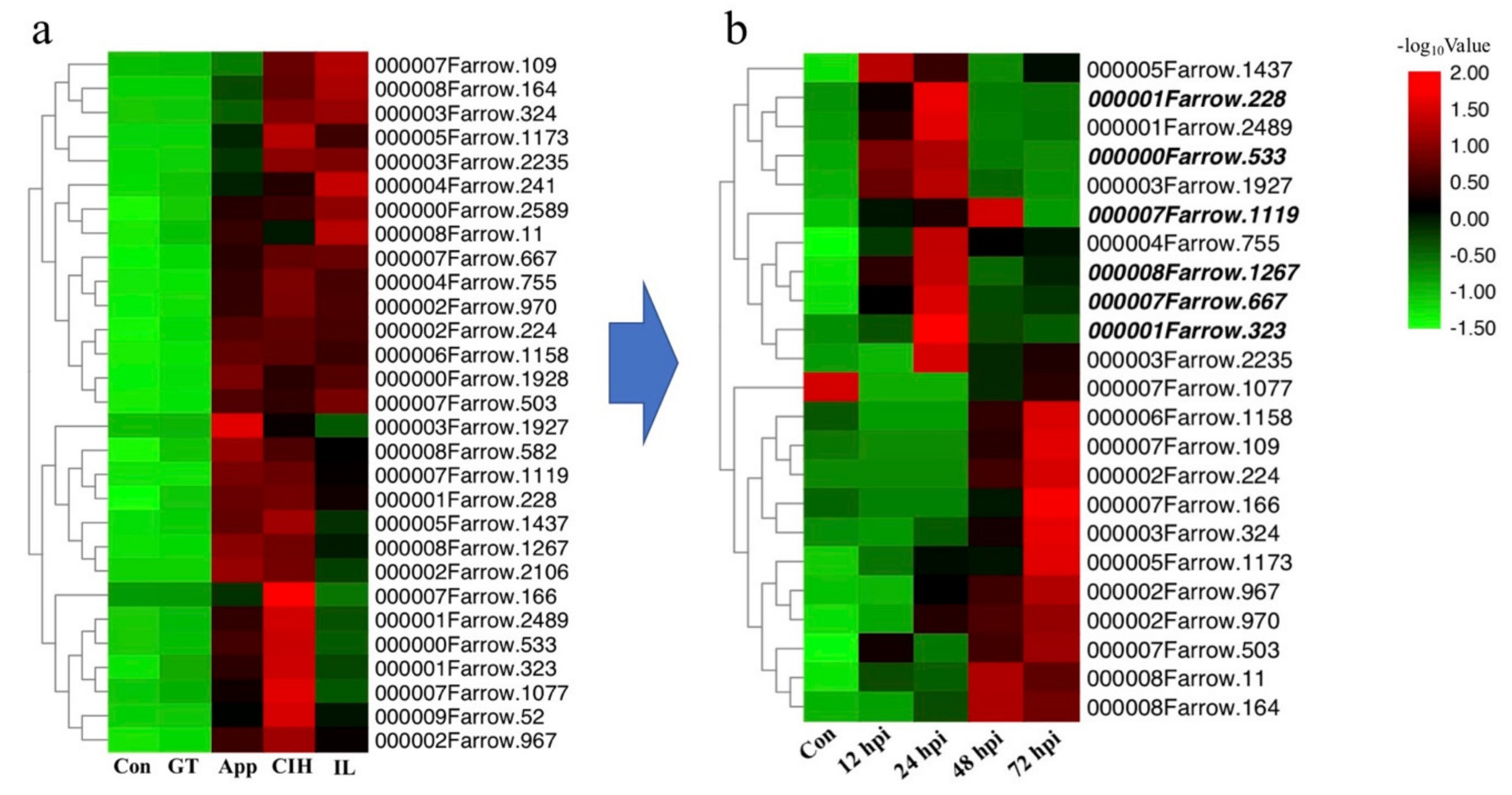
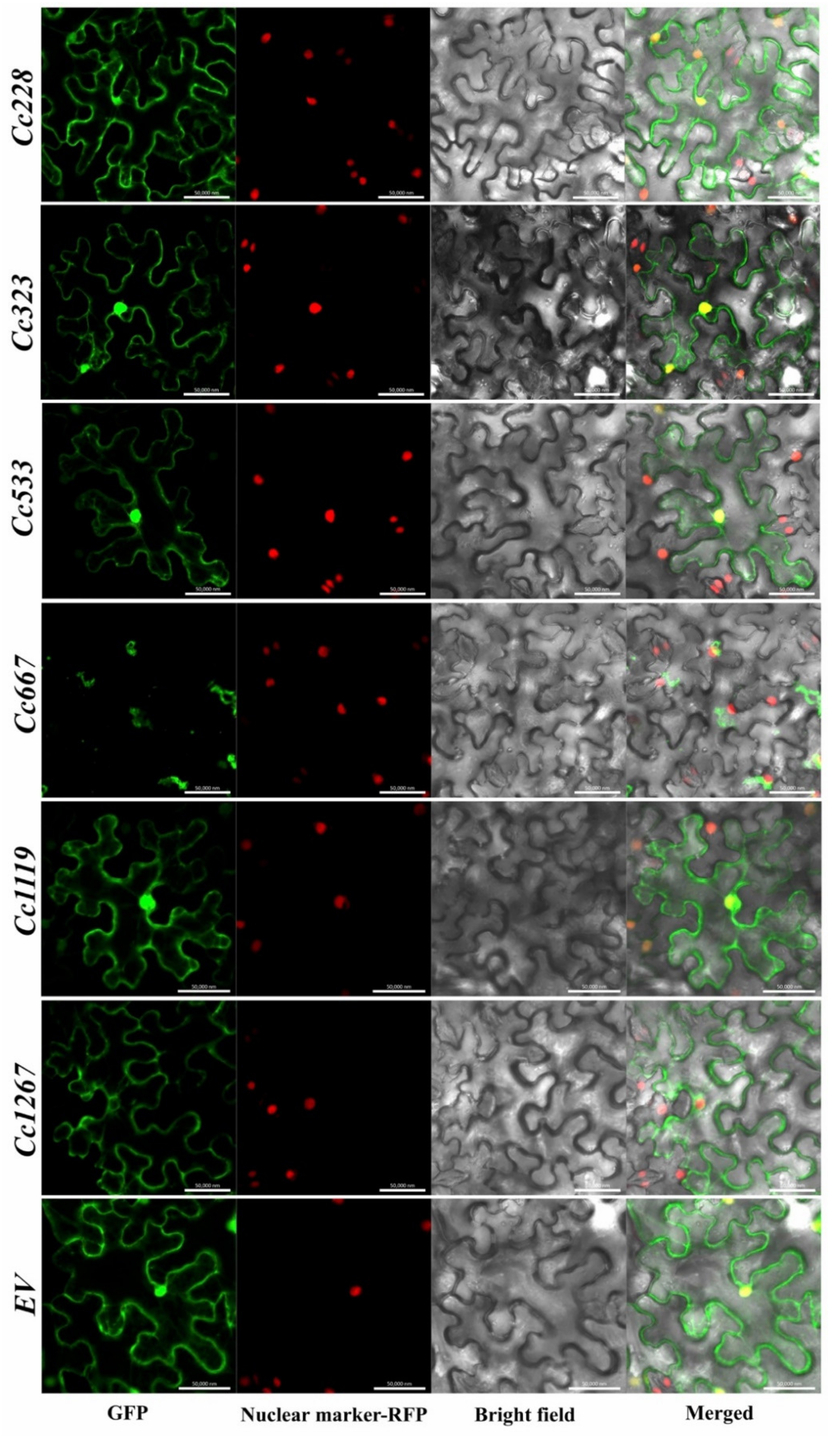
3.5. Prediction of Candidate Effector Proteins (CEPs) and Subcellular Localization Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Herb, B.; Richard, H.; Elizabeth, B.; Jenny, R.; Eleanor, L.; Nick, H. Tea. Nature 2019, 566, 7742. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Wang, Y.; Li, N.; Ni, D.; Yang, Y.; Wang, X. Differences in the characteristics and pathogenicity of Colletotrichum camelliae and C. fructicola isolated from the tea plant [Camellia sinensis (L.) O. Kuntze]. Front. Microbiol. 2018, 9, 3060. [Google Scholar] [CrossRef] [PubMed]
- Cannon, P.F.; Damm, U.; Johnston, P.R.; Weir, B.S. Colletotrichum - current status and future directions. Stud. Mycol. 2012, 73, 181–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Eliahu, N.; Igbaria, A.; Rose, M.S.; Horwitz, B.A.; Lev, S. Melanin biosynthesis in the maize pathogen Cochliobolus heterostrophus depends on two mitogen-activated protein kinases, Chk1 and Mps1, and the transcription factor Cmr1. Eukaryot. Cell 2007, 6, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latunde-Dada, A.O. Colletotrichum: Tales of forcible entry, stealth, transient confinement and breakout. Mol. Plant Pathol. 2001, 2, 187–198. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kawano, T.; Li, D.; Kolattukudy, P.E. A mitogen-activated protein kinase kinase required for induction of cytokinesis and appressorium formation by host signals in the conidia of Colletotrichum gloeosporioides. Plant Cell 2000, 12, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Okayama, H. A pcl-like cyclin activates the Res2p-Cdc10p cell cycle “start” transcriptional factor complex in fission yeast. Mol. Biol. Cell 2000, 11, 2845–2862. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhou, X.; Xu, J.R. Genetic control of infection-related development in Magnaporthe oryzae. Curr. Opin. Microbiol. 2012, 15, 678–684. [Google Scholar] [CrossRef]
- Shnaiderman, C.; Miyara, I.; Kobiler, I.; Sherman, A.; Prusky, D. Differential activation of ammonium transporters during the accumulation of ammonia by Colletotrichum gloeosporioides and its effect on appressoria formation and pathogenicity. Mol. Plant Microbe Interact. 2013, 26, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, R.J.; Valent, B. Breaking and entering: Host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu. Rev. Microbiol. 1996, 50, 491–512. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Alkan, N.; Friedlander, G.; Ment, D.; Prusky, D.; Fluhr, R. Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytol. 2015, 205, 801–815. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, X.; He, C.; Zhang, Q.Y.; Zou, X.; Duan, K.; Gao, Q. Novel fungal pathogenicity and leaf defense strategies are revealed by simultaneous transcriptome analysis of Colletotrichum fructicola and strawberry infected by this fungus. Front. Plant Sci. 2018, 9, 434. [Google Scholar] [CrossRef] [Green Version]
- Van den Brink, J.; de Vries, R.P. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477–1492. [Google Scholar] [CrossRef] [Green Version]
- Spadaro, D.; Droby, S. Development of biocontrol products for postharvest diseases of fruit: The importance of elucidating the mechanisms of action of yeast antagonists. Trends Food Sci. Technol. 2016, 47, 39–49. [Google Scholar] [CrossRef]
- Yoshida, K.; Ogino, A.; Yamada, K.; Sonoda, R. Induction of disease resistance in tea (Camellia sinensis L.) by plant activators. Jpn. Agric. Res. Q. 2010, 44, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Thomma, B.P.; Nurnberger, T.; Joosten, M.H. Of PAMPs and effectors: The blurred PTI-ETI dichotomy. Plant Cell 2011, 23, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Weir, B.S.; Damm, U.; Crous, P.W.; Wang, Y.; Liu, B.; Wang, M.; Zhang, M.; Cai, L. Unravelling Colletotrichum species associated with Camellia: Employing ApMat and GS loci to resolve species in the C. gloeosporioides complex. Persoonia 2015, 35, 63–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Shang, S.; Dong, Q.; Wang, B.; Zhang, R.; Gleason, M.L.; Sun, G. Transcriptomic analysis reveals candidate genes regulating development and host interactions of Colletotrichum fructicola. BMC Genomics 2018, 19, 557. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Hao, X.Y.; Wang, L.; Bin, X.; Wang, X.C.; Yang, Y.J. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 2016, 6, 35287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Chen, H.; Wei, Y.; An, T.; Liu, S. Development of a DNA-based real-time PCR assay for the quantification of Colletotrichum camelliae growth in tea (Camellia sinensis). Plant Methods 2020, 16, 17. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Yang, Y.; Yue, C.; Wang, L.; Horvath, D.P.; Wang, X. Comprehensive transcriptome analyses reveal differential gene expression profiles of Camellia sinensis axillary buds at para-, endo-, ecodormancy, and bud flush stages. Front. Plant Sci. 2017, 8, 553. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; An, T.; Runa, A.; Liu, S. Validation of reliable reference genes for RT-qPCR studies of target gene expression in Colletotrichum camelliae during spore germination and mycelial growth and interaction with host plants. Front. Microbiol. 2019, 10, 2055. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.; Talhinhas, P.; Loureiro, A.; Duplessis, S.; Fernandez, D.; Silva Mdo, C.; Paulo, O.S.; Azinheira, H.G. Validation of RT-qPCR reference genes for in planta expression studies in Hemileia vastatrix, the causal agent of coffee leaf rust. Fungal Biol. 2011, 115, 891–901. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Qi, M.; Mei, Y.; Grayczyk, J.P.; Darben, L.M.; Rieker, M.E.G.; Seitz, J.M.; Voegele, R.T.; Whitham, S.A.; Link, T.I. Candidate effectors from Uromyces appendiculatus, the causal agent of rust on common bean, can be discriminated based on suppression of immune responses. Front. Plant Sci. 2019, 10, 1182. [Google Scholar] [CrossRef] [Green Version]
- Robin, G.P.; Kleemann, J.; Neumann, U.; Cabre, L.; Dallery, J.-F.; Lapalu, N.; O’Connell, R.J. Subcellular localization screening of Colletotrichum higginsianum effector candidates identifies fungal proteins targeted to plant peroxisomes, golgi bodies, and microtubules. Front. Plant Sci. 2018, 9, 562. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Shim, S.H. The fungus Colletotrichum as a source for bioactive secondary metabolites. Arch. Pharm. Res. 2019, 42, 735–753. [Google Scholar] [CrossRef]
- Shao, Y.L.; Okuda, S.; Ikeda, K.; Okuno, T.; Takano, Y. LAC2 encoding a secreted laccase is involved in appressorial melanization and conidial pigmentation in Colletotrichum orbiculare. Mol. Plant Microbe Interact. 2012, 25, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Löhrer, M.; Hempel, M.; Mathea, S.; Schliebner, I.; Menzel, M.; Kiesow, A.; Schaffrath, U.; Deising, H.B.; Horbach, R. Melanin is not required for turgor generation but enhances cell-wall rigidity in appressoria of the corn pathogen Colletotrichum graminicola. Mol. Plant Microbe Interact. 2014, 27, 315–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Giraud, T.; Zhang, N.; Begerow, D.; Cai, G.; Shivas, R.G. The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers. 2011, 50, 121. [Google Scholar] [CrossRef]
- Hamer, J.E.; Howard, R.J.; Chumley, F.G.; Valent, B. A mechanism for surface attachment in spores of a plant pathogenic fungus. Science 1988, 239, 288–290. [Google Scholar] [CrossRef]
- Yan, Y.; Yuan, Q.; Tang, J.; Huang, J.; Hsiang, T.; Wei, Y.; Zheng, L. Colletotrichum higginsianum as a model for understanding host–Pathogen interactions: A review. Int. J. Mol. Sci. 2018, 19, 2142. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.; Tavares-Carreón, F.; Khodai-Kalaki, M.; Valvano, M.A.; Kivisaar, M. Tyrosine phosphorylation and dephosphorylation in Burkholderia cenocepacia affect biofilm formation, growth under nutritional deprivation, and pathogenicity. Appl. Environ. Microbiol. 2016, 82, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Doehlemann, G.; Ökmen, B.; Zhu, W.; Sharon, A. Plant pathogenic fungi. Microbiol. Spectr. 2017, 5, 1. [Google Scholar] [CrossRef]
- Wei, W.; Xiong, Y.; Zhu, W.; Wang, N.; Yang, G.; Peng, F. Colletotrichum higginsianum mitogen-activated protein kinase ChMK1: Role in growth, cell wall integrity, colony melanization, and pathogenicity. Front. Microbiol. 2016, 7, 1212. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, M.; Mukherjee, P.K.; Kale, S.P. cAMP signalling is involved in growth, germination, mycoparasitism and secondary metabolism in Trichoderma virens. Microbiology 2007, 153, 1734–1742. [Google Scholar] [CrossRef] [Green Version]
- Truesdell, G.M.; Yang, Z.; Dickman, M.B. A Gα subunit gene from the phytopathogenic fungus Colletotrichum trifolii is required for conidial germination. Physiol. Mol. Plant Pathol. 2000, 56, 131–140. [Google Scholar] [CrossRef]
- Takano, Y.; Kikuchi, T.; Kubo, Y.; Hamer, J.E.; Mise, K.; Furusawa, I. The Colletotrichum lagenarium MAP kinase gene CMK1 regulates diverse aspects of fungal pathogenesis. Mol. Plant Microbe Interact. 2000, 13, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.; Gong, X.; Hamid, M.I.; Fu, Y.; Jiatao, X.; Cheng, J.; Li, G.; Jiang, D. A fungal cell wall integrity-associated MAP kinase cascade in Coniothyrium minitans is required for conidiation and mycoparasitism. Fungal Genet. Biol. 2012, 49, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Villa-Rivera, M.G.; Conejo-Saucedo, U.; Lara-Marquez, A.; Cano-Camacho, H.; Lopez-Romero, E.; Zavala-Paramo, M.G. The role of virulence factors in the pathogenicity of Colletotrichum sp. Curr. Protein Pept. Sci. 2017, 18, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Bellincampi, D.; Cervone, F.; Lionetti, V. Plant cell wall dynamics and wall-related susceptibility in plant-pathogen interactions. Front. Plant Sci. 2014, 5, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Garcia, E.; Valent, B. How eukaryotic filamentous pathogens evade plant recognition. Curr. Opin. Microbiol. 2015, 26, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeson, W.T.; Phillips, C.M.; Cate, J.H.; Marletta, M.A. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 2012, 134, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.C.; Johansen, K.S.; Krogh, K.B.; Jørgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef] [Green Version]
- Kleemann, J.; Rincon-Rivera, L.J.; Takahara, H.; Neumann, U.; Ver Loren van Themaat, E.; van der Does, H.C.; Hacquard, S.; Stuber, K.; Will, I.; Schmalenbach, W.; et al. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathog. 2012, 8, e1002643. [Google Scholar] [CrossRef]
- Vogel, J. Unique aspects of the grass cell wall. Curr. Opin. Plant Biol. 2008, 11, 301–307. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, Y.; Du, Z.; Chen, X.; Zhou, X.; Kong, X.; Sun, W.; Chen, Z.; Chen, C.; Chen, M. Tender leaf and fully-expanded leaf exhibited distinct cuticle structure and wax lipid composition in Camellia sinensis cv Fuyun 6. Sci. Rep. 2018, 8, 14944. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Qian, W.J.; Li, N.N.; Hao, X.Y.; Wang, L.; Xiao, B.; Wang, X.C.; Yang, Y.J. Metabolic changes of caffeine in tea plant (Camellia sinensis (L.) O. Kuntze) as defense response to Colletotrichum fructicola. J. Agric. Food Chem. 2016, 64, 6685–6693. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, S.; Horwitz, B.A. Plant phenolic compounds and oxidative stress: Integrated signals in fungal-plant interactions. Curr. Genet. 2015, 61, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Azmi, N.S.A.; Singkaravanit-Ogawa, S.; Ikeda, K.; Kitakura, S.; Inoue, Y.; Narusaka, Y.; Shirasu, K.; Kaido, M.; Mise, K.; Takano, Y. Inappropriate expression of an NLP effector in Colletotrichum orbiculare impairs infection on cucurbitaceae cultivars via plant recognition of the C-terminal region. Mol. Plant Microbe Interact. 2018, 31, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Chen, S.; Shirsekar, G.; Zhou, B.; Khang, C.H.; Songkumarn, P.; Afzal, A.J.; Ning, Y.; Wang, R.; Bellizzi, M.; et al. The Magnaporthe oryzae effector AvrPiz-t targets the RING E3 ubiquitin ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in rice. Plant Cell 2012, 24, 4748–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Subcellular Localization |
|---|---|
| Cc228 | Nuclear + Cytoplasmic + Cytomembrane |
| Cc323 | Nuclear + Cytoplasmic + Cytomembrane |
| Cc533 | Nuclear + Cytoplasmic + Cytomembrane |
| Cc667 | Cytoplasmic |
| Cc1119 | Nuclear + Cytoplasmic + Cytomembrane |
| Cc1267 | Karyolemma + Cytoplasmic + Cytomembrane |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, F.; Wang, Y.; Lu, Q.; Hao, X.; Fang, W.; Yang, Y.; Zhu, X.; Wang, X. Lifestyle Characteristics and Gene Expression Analysis of Colletotrichum camelliae Isolated from Tea Plant [Camellia sinensis (L.) O. Kuntze] Based on Transcriptome. Biomolecules 2020, 10, 782. https://doi.org/10.3390/biom10050782
Xiong F, Wang Y, Lu Q, Hao X, Fang W, Yang Y, Zhu X, Wang X. Lifestyle Characteristics and Gene Expression Analysis of Colletotrichum camelliae Isolated from Tea Plant [Camellia sinensis (L.) O. Kuntze] Based on Transcriptome. Biomolecules. 2020; 10(5):782. https://doi.org/10.3390/biom10050782
Chicago/Turabian StyleXiong, Fei, Yuchun Wang, Qinhua Lu, Xinyuan Hao, Wanping Fang, Yajun Yang, Xujun Zhu, and Xinchao Wang. 2020. "Lifestyle Characteristics and Gene Expression Analysis of Colletotrichum camelliae Isolated from Tea Plant [Camellia sinensis (L.) O. Kuntze] Based on Transcriptome" Biomolecules 10, no. 5: 782. https://doi.org/10.3390/biom10050782