The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases

 ,
,  ,
,  , and
, and
Abstract
:1. AKI, CKD and the Mitochondrial Connection
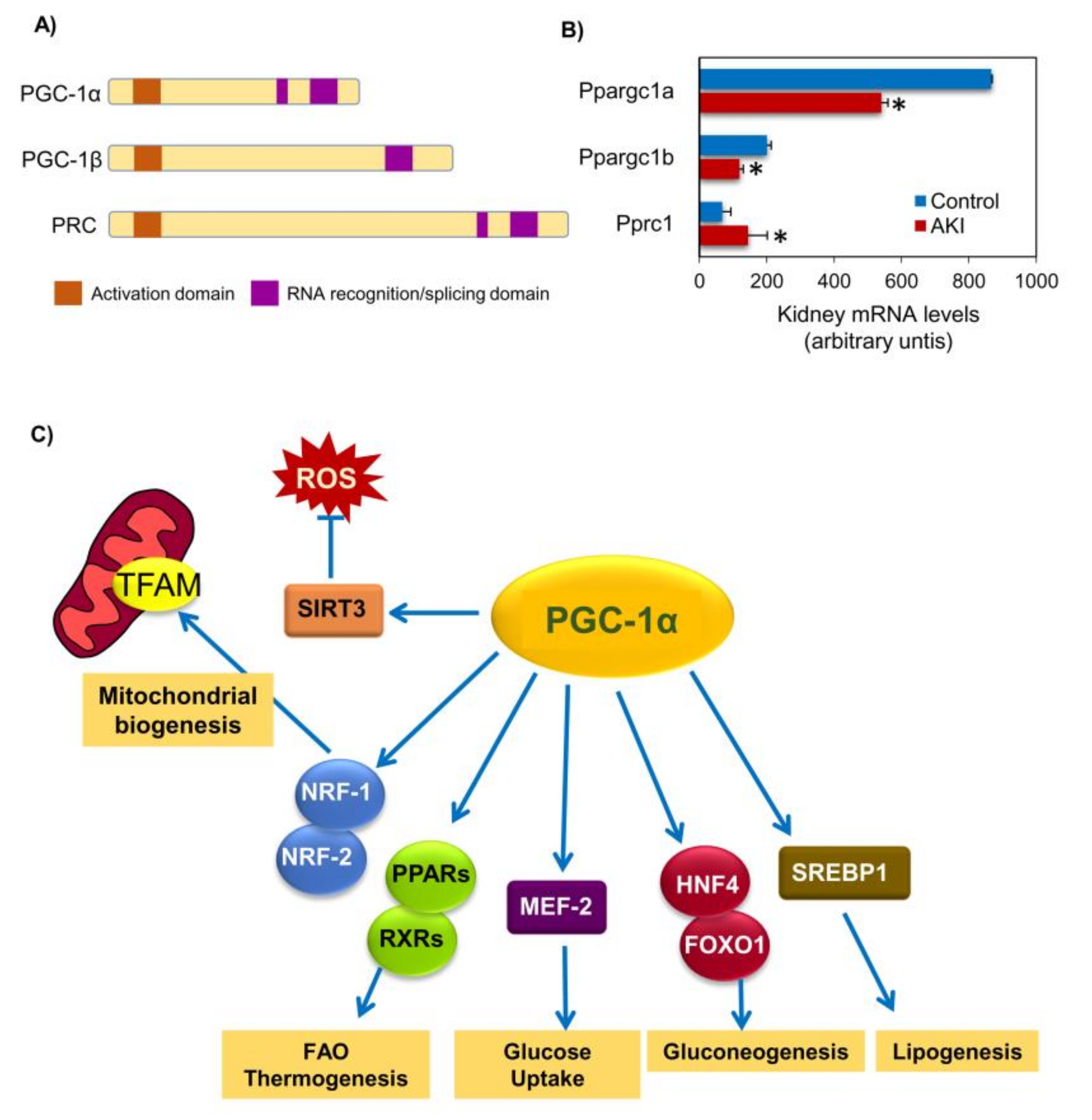
2. PGC-1α: A Regulator of Mitochondrial Biogenesis
3. PGC-1α in Health and Disease
3.1. Diabetes
3.2. Pancreatitis
3.3. Liver Disease
3.4. Endothelium
4. Mitochondria in Kidney Diseases
4.1. Morphological and Functional Changes of Mitochondria
4.2. Mitochondria-Targeted Therapies
5. PGC-1α in Kidney Diseases
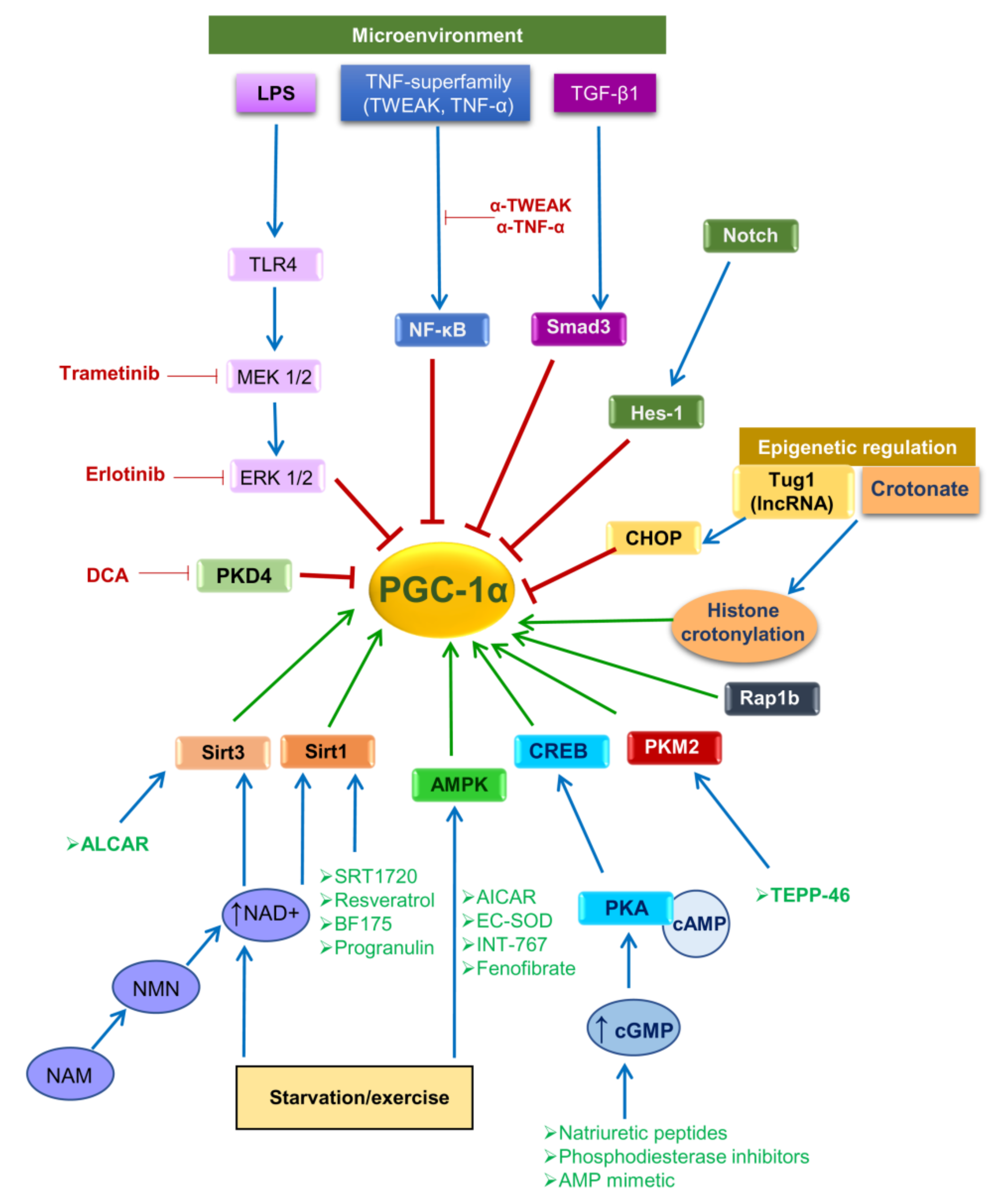
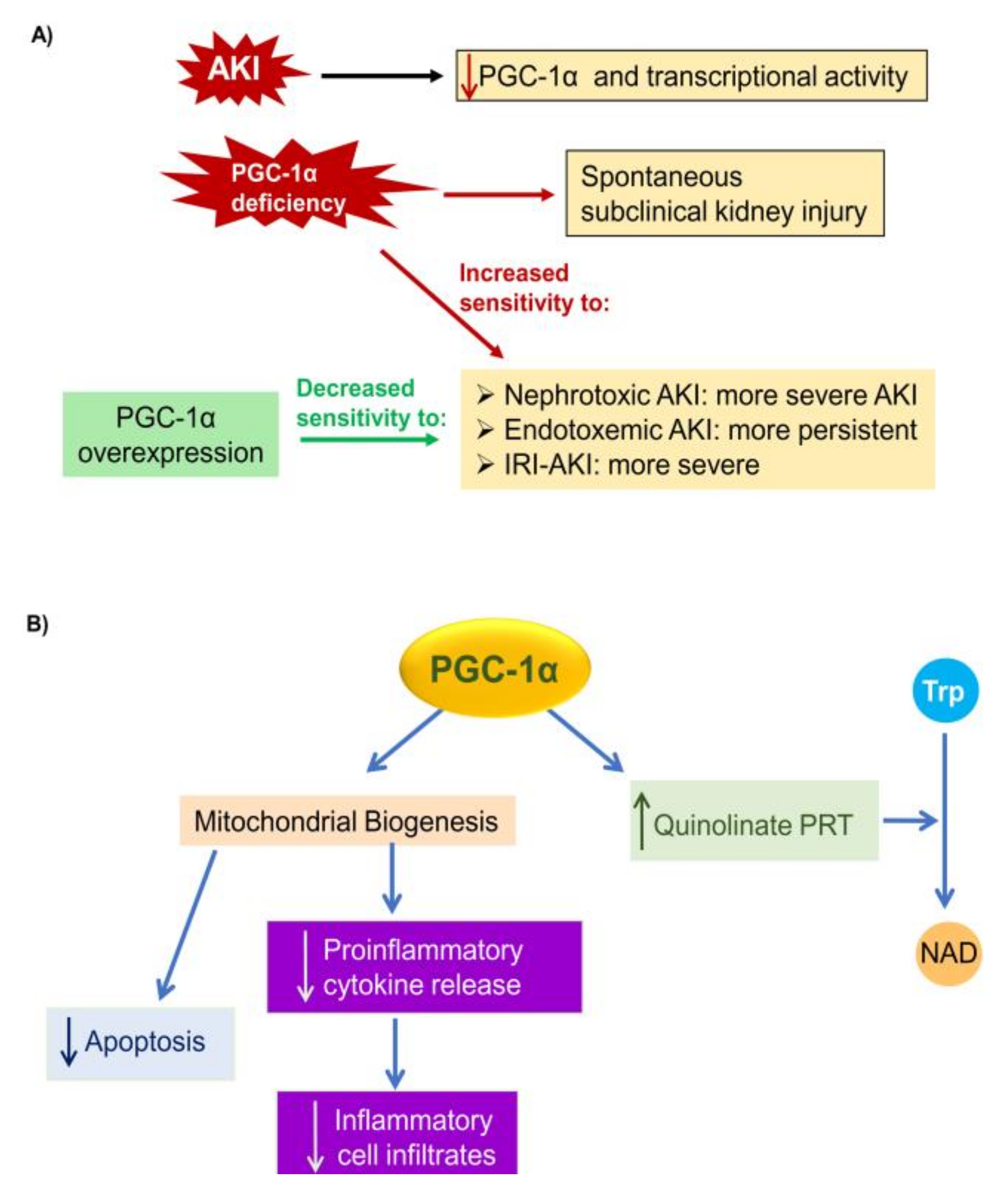
5.1. PGC-1α in AKI
5.2. PGC-1α in CKD
6. Summary and Future Perspectives
Funding
Conflicts of Interest
References
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Iavecchia, L.; Cereza García, G.; Sabaté Gallego, M.; Vidal Guitart, X.; Ramos Terrades, N.; de la Torre, J.; Segarra Medrano, A.; Agustí Escasany, A. Drug-related acute renal failure in hospitalised patients. Nefrologia 2015, 35, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo, E.; Suberviola, B.; Albines, Z.; Castellanos, Á.; Heras, M.; Rodriguez-Borregán, J.C.; Piñera, C.; Serrano, M.; Arias, M. A comparison of acute kidney injury classification systems in sepsis. Nefrologia 2016, 36, 530–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol 2017, 28, 218–229. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; Mässenhausen, A.V.; Linkermann, A.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. USA 2018, 115, 4182–4187. [Google Scholar] [CrossRef] [Green Version]
- Von Mässenhausen, A.; Tonnus, W.; Linkermann, A. Cell Death Pathways Drive Necroinflammation during Acute Kidney Injury. Nephron 2018, 140, 144–147. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Poveda, J.; Fontecha-Barriuso, M.; Ruiz-Andres, O.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting of regulated necrosis in kidney disease. Nefrologia 2018, 38, 125–135. [Google Scholar] [CrossRef]
- Sanz, A.B.; Santamaría, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 2008, 19, 1634–1642. [Google Scholar] [CrossRef] [Green Version]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Sanchez-Niño, M.D.; Crespo-Barrio, M.; De-Sequera-Ortiz, P.; Fernández-Giráldez, E.; García-Maset, R.; Macía-Heras, M.; Pérez-Fontán, M.; Rodríguez-Portillo, M.; Salgueira-Lazo, M.; et al. The Spanish Society of Nephrology (SENEFRO) commentary to the Spain GBD 2016 report: Keeping chronic kidney disease out of sight of health authorities will only magnify the problem. Nefrologia 2019, 39, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Inter. 2013, 3, 1–150. [Google Scholar]
- Perez-Gomez, M.V.; Bartsch, L.A.; Castillo-Rodriguez, E.; Fernandez-Prado, R.; Fernandez-Fernandez, B.; Martin-Cleary, C.; Gracia-Iguacel, C.; Ortiz, A. Clarifying the concept of chronic kidney disease for non-nephrologists. Clin. Kidney J. 2019, 12, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Hsu, R.K.; Hsu, C.Y. The Role of Acute Kidney Injury in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 283–292. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Honarpisheh, M.; Desai, J.; Marschner, J.A.; Weidenbusch, M.; Lech, M.; Vielhauer, V.; Anders, H.J.; Mulay, S.R. Regulated necrosis-related molecule mRNA expression in humans and mice and in murine acute tissue injury and systemic autoimmunity leading to progressive organ damage, and progressive fibrosis. Biosci. Rep. 2016, 36, e00425. [Google Scholar] [CrossRef]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Ortiz, A.; Sanchez-Niño, M.D. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, M.A.; Weinberg, J.M. The tubule pathology of septic acute kidney injury: A neglected area of research comes of age. Kidney Int. 2012, 81, 338–340. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Nuñez, G.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol. Dial. Transplant. 2019. [Google Scholar] [CrossRef]
- Valiño-Rivas, L.; Cuarental, L.; Agustin, M.; Husi, H.; Cannata-Ortiz, P.; Sanz, A.B.; Mischak, H.; Ortiz, A.; Sanchez-Niño, M.D. MAGE genes in the kidney: Identification of MAGED2 as upregulated during kidney injury and in stressed tubular cells. Nephrol. Dial. Transplant. 2019, 34, 1498–1507. [Google Scholar] [CrossRef]
- Poveda, J.; Sanz, A.B.; Carrasco, S.; Ruiz-Ortega, M.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ortiz, A. Bcl3: A regulator of NF-κB inducible by TWEAK in acute kidney injury with anti-inflammatory and antiapoptotic properties in tubular cells. Exp. Mol. Med. 2017, 49, e352. [Google Scholar] [CrossRef] [Green Version]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [Green Version]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Ann. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Wills, L.P.; Stallons, L.J.; Schnellmann, R.G. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 347, 626–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Yuan, Y.; Li, S.; Zeng, C.; Yu, W.; Shen, M.; Zhang, R.; Li, C.; Zhang, Y.; Wang, H. PDE5 inhibitors protect against post-infarction heart failure. Front. Biosci. 2016, 21, 1194–1210. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sánchez-Ramos, C.; Monsalve, M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Hindi, S.M.; Mishra, V.; Bhatnagar, S.; Tajrishi, M.M.; Ogura, Y.; Yan, Z.; Burkly, L.C.; Zheng, T.S.; Kumar, A. Regulatory circuitry of TWEAK-Fn14 system and PGC-1α in skeletal muscle atrophy program. FASEB J. 2014, 28, 1398–1411. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1α Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Susztak, K. The Role of Peroxisome Proliferator-Activated Receptor γ Coactivator 1α (PGC-1α) in Kidney Disease. Semin. Nephrol. 2018, 38, 121–126. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): Requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Xu, G.; Deeney, J.T.; Yang, S.N.; Rhee, J.; Puigserver, P.; Levens, A.R.; Yang, R.; Zhang, C.Y.; Lowell, B.B.; et al. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev. Cell. 2003, 5, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.S.; Jun, H.S. Role of bioactive food components in diabetes prevention: Effects on Beta-cell function and preservation. Nutr. Metab. Insights 2014, 7, 51–59. [Google Scholar] [CrossRef]
- De Souza, C.T.; Gasparetti, A.L.; Pereira-da-Silva, M.; Araújo, E.P.; Carvalheira, J.B.; Saad, M.J.; Boschero, A.C.; Carneiro, E.M.; Velloso, L.A. Peroxisome proliferator-activated receptor gamma coactivator-1-dependent uncoupling protein-2 expression in pancreatic islets of rats: A novel pathway for neural control of insulin secretion. Diabetologia 2003, 46, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.L.; Jiang, B.G.; Li, W.T.; Zou, J.J.; Shi, Y.Q.; Liu, Z.M. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res. Clin. Pract. 2011, 91, 94–100. [Google Scholar] [CrossRef]
- Kim, J.W.; Sun, C.; Jeon, S.Y.; You, Y.H.; Shin, J.Y.; Lee, S.H.; Cho, J.H.; Park, C.G.; Yoon, K.H. Glucocorticoid treatment independently affects expansion and transdifferentiation of porcine neonatal pancreas cell clusters. BMB Rep. 2012, 45, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Park, S.Y.; You, Y.H.; Ham, D.S.; Park, H.S.; Lee, S.H.; Yang, H.K.; Yoon, K.H. Targeting PGC-1α to overcome the harmful effects of glucocorticoids in porcine neonatal pancreas cell clusters. Transplantation 2014, 97, 273–279. [Google Scholar] [CrossRef]
- Vandenbeek, R.; Khan, N.P.; Estall, J.L. Linking Metabolic Disease With the PGC-1α Gly482Ser Polymorphism. Endocrinology 2018, 159, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Del Guerra, S.; Lupi, R.; Rönn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; Del Prato, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyenechea, E.; Crujeiras, A.B.; Abete, I.; Parra, D.; Martínez, J.A. Enhanced short-term improvement of insulin response to a low-caloric diet in obese carriers the Gly482Ser variant of the PGC-1alpha gene. Diabetes Res. Clin. Pract. 2008, 82, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, C.; Zhang, C.; Zhang, Y.; Shen, P.; Zhang, J.; Zhang, C.Y. Free fatty acids increase PGC-1alpha expression in isolated rat islets. FEBS Lett. 2005, 579, 1446–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.T.; Cao, X.P.; Chen, R.Z.; Zhu, X.N.; Wang, X.L.; Li, Y.B.; Xiao, H.P. Down-regulation of peroxisome proliferator-activated receptor γ coactivator-1α expression in fatty acid-induced pancreatic beta-cell apoptosis involves nuclear factor-κB pathway. Chin. Med. J. 2011, 124, 3657–3663. [Google Scholar] [PubMed]
- Oropeza, D.; Jouvet, N.; Bouyakdan, K.; Perron, G.; Ringuette, L.J.; Philipson, L.H.; Kiss, R.S.; Poitout, V.; Alquier, T.; Estall, J.L. PGC-1 coactivators in β-cells regulate lipid metabolism and are essential for insulin secretion coupled to fatty acids. Mol. Metab. 2015, 4, 811–822. [Google Scholar] [CrossRef]
- Kramer, A.; Pippias, M.; Noordzij, M.; Stel, V.S.; Andrusev, A.M.; Aparicio-Madre, M.I.; Arribas Monzón, F.E.; Åsberg, A.; Barbullushi, M.; Beltrán, P.; et al. The European Renal Association - European Dialysis and Transplant Association (ERA-EDTA) Registry Annual Report 2016: A summary. Clin. Kidney J. 2019, 12, 702–720. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Li, L.; Wang, C.; Yang, H.; Liu, S.; Lu, Y.; Fu, P.; Liu, J. Metabolomics reveal mitochondrial and fatty acid metabolism disorders that contribute to the development of DKD in T2DM patients. Mol. Biosyst. 2017, 13, 2392–2400. [Google Scholar] [CrossRef]
- Pérez, S.; Rius-Pérez, S.; Finamor, I.; Martí-Andrés, P.; Prieto, I.; García, R.; Monsalve, M.; Sastre, J. Obesity causes PGC-1α deficiency in the pancreas leading to marked IL-6 upregulation via NF-κB in acute pancreatitis. J. Pathol. 2019, 247, 48–59. [Google Scholar] [CrossRef]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jäger, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Invest 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Vodkin, I.; Kuo, A. Extended Criteria Donors in Liver Transplantation. Clin. Liver Dis. 2017, 21, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ramos, C.; Prieto, I.; Tierrez, A.; Laso, J.; Valdecantos, M.P.; Bartrons, R.; Roselló-Catafau, J.; Monsalve, M. PGC-1α Downregulation in Steatotic Liver Enhances Ischemia-Reperfusion Injury and Impairs Ischemic Preconditioning. Antioxid. Redox Signal. 2017, 27, 1332–1346. [Google Scholar] [CrossRef] [Green Version]
- Chaung, W.W.; Jacob, A.; Ji, Y.; Wang, P. Suppression of PGC-1alpha by Ethanol: Implications of Its Role in Alcohol Induced Liver Injury. Int. J. Clin. Exp. Med. 2008, 1, 161–170. [Google Scholar]
- Zhang, X.; Ji, R.; Sun, H.; Peng, J.; Ma, X.; Wang, C.; Fu, Y.; Bao, L.; Jin, Y. Scutellarin ameliorates nonalcoholic fatty liver disease through the PPARγ/PGC-1α-Nrf2 pathway. Free Radic. Res. 2018, 52, 198–211. [Google Scholar] [CrossRef]
- Yao, W.; Cai, H.; Li, X.; Li, T.; Hu, L.; Peng, T. Endoplasmic reticulum stress links hepatitis C virus RNA replication to wild-type PGC-1α/liver-specific PGC-1α upregulation. J. Virol. 2014, 88, 8361–8374. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Tang, C.; Zhang, Y.; Cheng, Y.; Cai, L.; Chen, X.; Gao, Y.; Deng, Y.; Pan, M. SIRT1/PGC-1α signaling protects hepatocytes against mitochondrial oxidative stress induced by bile acids. Free Radic. Res. 2015, 49, 935–945. [Google Scholar] [CrossRef]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef]
- Kim, M.S.; Sweeney, T.R.; Shigenaga, J.K.; Chui, L.G.; Moser, A.; Grunfeld, C.; Feingold, K.R. Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1alpha, and PGC-1beta in liver cells. Metabolism 2007, 56, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speckmann, B.; Walter, P.L.; Alili, L.; Reinehr, R.; Sies, H.; Klotz, L.O.; Steinbrenner, H. Selenoprotein P expression is controlled through interaction of the coactivator PGC-1alpha with FoxO1a and hepatocyte nuclear factor 4alpha transcription factors. Hepatology 2008, 48, 1998–2006. [Google Scholar] [CrossRef]
- Lopes Junior, E.; Leite, H.P.; Konstantyner, T. Selenium and selenoproteins: From endothelial cytoprotection to clinical outcomes. Transl. Res. 2019, 208, 85–104. [Google Scholar] [CrossRef]
- Felder, T.K.; Soyal, S.M.; Oberkofler, H.; Hahne, P.; Auer, S.; Weiss, R.; Gadermaier, G.; Miller, K.; Krempler, F.; Esterbauer, H.; et al. Characterization of novel peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) isoform in human liver. J. Biol. Chem. 2011, 286, 42923–42936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, C.D.; Targher, G. NAFLD as a driver of chronic kidney disease. J. Hepatol. 2020, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattacheril, J. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gastroenterol. Clin. 2020, 49, 141–149. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006, 20, 1889–1891. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Zhou, Y.; Cai, W.; Qiu, L. C1q/TNF-Related Protein-9 Ameliorates Ox-LDL-Induced Endothelial Dysfunction via PGC-1α/AMPK-Mediated Antioxidant Enzyme Induction. Int. J. Mol. Sci. 2017, 18, e1097. [Google Scholar] [CrossRef] [Green Version]
- García-Quintans, N.; Prieto, I.; Sánchez-Ramos, C.; Luque, A.; Arza, E.; Olmos, Y.; Monsalve, M. Regulation of endothelial dynamics by PGC-1α relies on ROS control of VEGF-A signaling. Free Radic. Biol. Med. 2016, 93, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Borniquel, S.; García-Quintáns, N.; Valle, I.; Olmos, Y.; Wild, B.; Martínez-Granero, F.; Soria, E.; Lamas, S.; Monsalve, M. Inactivation of Foxo3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Mol. Cell. Biol. 2010, 30, 4035–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Quintans, N.; Sánchez-Ramos, C.; Prieto, I.; Tierrez, A.; Arza, E.; Alfranca, A.; Redondo, J.M.; Monsalve, M. Oxidative stress induces loss of pericyte coverage and vascular instability in PGC-1α-deficient mice. Angiogenesis 2016, 19, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model. Mech. 2018, 11, dmm032698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whaley-Connell, A.; Sowers, J.R. Obesity and kidney disease: From population to basic science and the search for new therapeutic targets. Kidney Int. 2017, 92, 313–323. [Google Scholar] [CrossRef]
- Zannad, F.; Rossignol, P. Cardiorenal Syndrome Revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Vanholder, R.; Fouque, D.; Glorieux, G.; Heine, G.H.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Ortiz, A.; Rossignol, P.; Wiecek, A.; et al. Clinical management of the uraemic syndrome in chronic kidney disease. Lancet Diabetes Endocrinol. 2016, 4, 360–373. [Google Scholar] [CrossRef]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef]
- Soltoff, S.P. ATP and the regulation of renal cell function. Ann. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef]
- Weidemann, M.J.; Krebs, H.A. The fuel of respiration of rat kidney cortex. Biochem. J. 1969, 112, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Ann. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell. Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.M.; Gomez, H.; Collage, R.D.; Loughran, P.; Zhang, X.; Escobar, D.A.; Billiar, T.R.; Zuckerbraun, B.S.; Rosengart, M.R. Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS ONE 2013, 8, e69520. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [Green Version]
- Galloway, C.A.; Lee, H.; Nejjar, S.; Jhun, B.S.; Yu, T.; Hsu, W.; Yoon, Y. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes 2012, 61, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [Green Version]
- Bartz, R.R.; Fu, P.; Suliman, H.B.; Crowley, S.D.; MacGarvey, N.C.; Welty-Wolf, K.; Piantadosi, C.A. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS ONE 2014, 9, e100912. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Jankauskas, S.S.; Andrianova, N.V.; Alieva, I.B.; Prusov, A.N.; Matsievsky, D.D.; Zorova, L.D.; Pevzner, I.B.; Savchenko, E.S.; Pirogov, Y.A.; Silachev, D.N.; et al. Dysfunction of Kidney Endothelium after Ischemia/Reperfusion and Its Prevention by Mitochondria-Targeted Antioxidant. Biochemistry 2016, 81, 1538–1548. [Google Scholar] [CrossRef]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorova, L.V.; Sodhi, K.; Gatto-Weis, C.; Puri, N.; Hinds, T.D.; Shapiro, J.I.; Malhotra, D. Peroxisome proliferator-activated receptor δ agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS ONE 2013, 8, e64436. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Kazachenko, A.V.; Vyssokikh, M.Y.; Vasileva, A.K.; Tcvirkun, D.V.; Isaev, N.K.; Kirpatovsky, V.I.; Zorov, D.B. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007, 72, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Heidari, R.; Ahmadi, A.; Mohammadi, H.; Ommati, M.M.; Azarpira, N.; Niknahad, H. Mitochondrial dysfunction and oxidative stress are involved in the mechanism of methotrexate-induced renal injury and electrolytes imbalance. Biomed. Pharmacother. 2018, 107, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Niknahad, H.; Heidari, R.; Mohammadzadeh, R.; Ommati, M.M.; Khodaei, F.; Azarpira, N.; Abdoli, N.; Zarei, M.; Asadi, B.; Rasti, M.; et al. Sulfasalazine induces mitochondrial dysfunction and renal injury. Ren. Fail. 2017, 39, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Horváth, B.; Zsengellér, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [Green Version]
- López, S.; Negredo, E.; Garrabou, G.; Puig, J.; Ruiz, L.; Sanjurjo, E.; Ramos, X.; Infante, A.B.; Casademont, J.; Cardellach, F.; et al. Longitudinal study on mitochondrial effects of didanosine-tenofovir combination. AIDS Res. Hum. Retroviruses 2006, 22, 33–39. [Google Scholar] [CrossRef]
- Yuan, J.; Zhou, J.; Chen, B.C.; Zhang, X.; Zhou, H.M.; Du, D.F.; Chang, S.; Chen, Z.K. Magnesium supplementation prevents chronic cyclosporine nephrotoxicity via adjusting nitric oxide synthase activity. Transplant. Proc. 2005, 37, 1892–1895. [Google Scholar] [CrossRef]
- Zsengellér, Z.K.; Ellezian, L.; Brown, D.; Horváth, B.; Mukhopadhyay, P.; Kalyanaraman, B.; Parikh, S.M.; Karumanchi, S.A.; Stillman, I.E.; Pacher, P. Cisplatin nephrotoxicity involves mitochondrial injury with impaired tubular mitochondrial enzyme activity. J. Histochem. Cytochem. 2012, 60, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Y.; Wang, Q.; Yang, J.; Yang, D.; Liu, J.; Li, J. Involvement of mitochondria-mediated apoptosis in ethylbenzene-induced renal toxicity in rat. Toxicol. Sci. 2010, 115, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Mitochondriopathies. Eur. J. Neurol. 2004, 11, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Goto, Y.; Nagata, M.; Yamaguchi, Y. Granular swollen epithelial cells: A histologic and diagnostic marker for mitochondrial nephropathy. Am. J. Surg. Pathol. 2010, 34, 262–270. [Google Scholar] [CrossRef]
- Kawakami, T.; Gomez, I.G.; Ren, S.; Hudkins, K.; Roach, A.; Alpers, C.E.; Shankland, S.J.; D’Agati, V.D.; Duffield, J.S. Deficient Autophagy Results in Mitochondrial Dysfunction and FSGS. J. Am. Soc. Nephrol. 2015, 26, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Itami, N.; Kajii, N.; Tochimaru, H.; Endo, M.; Horai, S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J. Pediatr. 1990, 116, 904–910. [Google Scholar] [CrossRef]
- Lake, N.J.; Bird, M.J.; Isohanni, P.; Paetau, A. Leigh syndrome: Neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 2015, 74, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Xie, P.; Wada, J.; Kashihara, N.; Liu, F.Y.; Zhao, Y.; Kumar, D.; Chugh, S.S.; Danesh, F.R.; Kanwar, Y.S. Rap1b GTPase ameliorates glucose-induced mitochondrial dysfunction. J. Am. Soc. Nephrol. 2008, 19, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.L.; Sourris, K.C.; Harcourt, B.E.; Thallas-Bonke, V.; Penfold, S.; Andrikopoulos, S.; Thomas, M.C.; O’Brien, R.C.; Bierhaus, A.; Cooper, M.E.; et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2010, 298, F763–F770. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef]
- Zepeda-Orozco, D.; Kong, M.; Scheuermann, R.H. Molecular Profile of Mitochondrial Dysfunction in Kidney Transplant Biopsies Is Associated With Poor Allograft Outcome. Transplant. Proc. 2015, 47, 1675–1682. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Bennett, N.C.; Roy, S.; Gabrielli, B.G.; Johnson, D.W.; Gobe, G.C. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron. Exp. Nephrol. 2012, 122, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shi, S.; Sun, X.; Cai, G.; Cui, S.; Hong, Q.; Chen, X.; Bai, X.Y. Mitochondrial autophagy involving renal injury and aging is modulated by caloric intake in aged rat kidneys. PLoS ONE 2013, 8, e69720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukunyadzi, P.; Huang, H.; Liu, K.; Fan, C.Y. Concomitant loss of mitochondria and the DNA repair protein hOGG1 in clear cell carcinoma of the kidney. Appl. Immunohistochem. Mol. Morphol. 2003, 11, 334–338. [Google Scholar] [CrossRef]
- Hervouet, E.; Godinot, C. Mitochondrial disorders in renal tumors. Mitochondrion 2006, 6, 105–117. [Google Scholar] [CrossRef]
- Hervouet, E.; Simonnet, H.; Godinot, C. Mitochondria and reactive oxygen species in renal cancer. Biochimie 2007, 89, 1080–1088. [Google Scholar] [CrossRef]
- Meierhofer, D.; Mayr, J.A.; Foetschl, U.; Berger, A.; Fink, K.; Schmeller, N.; Hacker, G.W.; Hauser-Kronberger, C.; Kofler, B.; Sperl, W. Decrease of mitochondrial DNA content and energy metabolism in renal cell carcinoma. Carcinogenesis 2004, 25, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Lagler, F.B. Current and Emerging Therapies for Mitochondriopathies. Handb. Exp. Pharmacol. 2019, 1–9. [Google Scholar] [CrossRef]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Liu, S.; Soong, Y.; Szeto, H.H. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim. Biophys. Acta 2015, 1847, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, H.H. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J. Am. Soc. Nephrol. 2017, 28, 2856–2865. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tang, D.; Zou, Y.; Wu, X.; Chen, Y.; Li, H.; Chen, S.; Shi, Y.; Niu, H. A mitochondrial-targeted peptide ameliorated podocyte apoptosis through a HOCl-alb-enhanced and mitochondria-dependent signalling pathway in diabetic rats and in vitro. J. Enzym. Inhib. Med. Chem. 2019, 34, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [Green Version]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2(+/)⁻(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 4, 121ra118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierro-Fernández, M.; Miguel, V.; Márquez-Expósito, L.; Nuevo-Tapioles, C.; Herrero, J.I.; Blanco-Ruiz, E.; Tituaña, J.; Castillo, C.; Cannata, P.; Monsalve, M.; et al. MiR-9-5p protects from kidney fibrosis by metabolic reprogramming. FASEB J. 2020, 34, 410–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Rasbach, K.A.; Schnellmann, R.G. Signaling of mitochondrial biogenesis following oxidant injury. J. Biol. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.A.; Gattone, V.H. Mitochondrial alterations in cisplatin-induced acute renal failure. Am. J. Physiol. 1986, 250, F991–F998. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Liu, F.; Dong, Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch. Toxicol. 2014, 88, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Santos, N.A.; Catão, C.S.; Martins, N.M.; Curti, C.; Bianchi, M.L.; Santos, A.C. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Arch. Toxicol. 2007, 81, 495–504. [Google Scholar] [CrossRef]
- Ahmed, L.A.; Shehata, N.I.; Abdelkader, N.F.; Khattab, M.M. Tempol, a superoxide dismutase mimetic agent, ameliorates cisplatin-induced nephrotoxicity through alleviation of mitochondrial dysfunction in mice. PLoS ONE 2014, 9, e108889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Inoue, K.; Matsumoto, T.; Ishihara, M.; Hamada, K.; Shimamura, Y.; Ogata, K.; Taniguchi, Y.; Horino, T.; Karashima, T.; et al. 5-Aminolevulinic acid protects against cisplatin-induced nephrotoxicity without compromising the anticancer efficiency of cisplatin in rats in vitro and in vivo. PLoS ONE 2013, 8, e80850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.J.; Ha, C.M.; Choi, Y.K.; Park, S.; Choe, M.S.; Jeoung, N.H.; Huh, Y.H.; Kim, H.J.; Kweon, H.S.; Lee, J.M.; et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017, 91, 880–895. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [Green Version]
- Oh, G.S.; Kim, H.J.; Choi, J.H.; Shen, A.; Choe, S.K.; Karna, A.; Lee, S.H.; Jo, H.J.; Yang, S.H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD+ levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ 2019, 364, k4891. [Google Scholar] [CrossRef]
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Stallons, L.J.; Collier, J.B.; Chavin, K.D.; Schnellmann, R.G. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2015, 352, 346–357. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, J.; Tian, J.; Virzì, G.M.; Digvijay, K.; Cueto, L.; Yin, Y.; Rosner, M.H.; Ronco, C. Mitochondria in Sepsis-Induced AKI. J. Am. Soc. Nephrol. 2019, 30, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jiang, B.; Qiu, Y.; Guan, J.; Jain, M.; Cao, X.; Bauer, M.; Su, L.; Burkly, L.C.; Leone, T.C.; et al. PGC1α plays a critical role in TWEAK-induced cardiac dysfunction. PLoS ONE 2013, 8, e54054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, T.; Jové, M.; Rodríguez-Calvo, R.; Eyre, E.; Palomer, X.; Sánchez, R.M.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes 2006, 55, 2779–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashabi, G.; Ramin, M.; Azizi, P.; Taslimi, Z.; Alamdary, S.Z.; Haghparast, A.; Ansari, N.; Motamedi, F.; Khodagholi, F. ERK and p38 inhibitors attenuate memory deficits and increase CREB phosphorylation and PGC-1α levels in Aβ-injected rats. Behav. Brain Res. 2012, 232, 165–173. [Google Scholar] [CrossRef]
- Khader, A.; Yang, W.L.; Hansen, L.W.; Rajayer, S.R.; Prince, J.M.; Nicastro, J.M.; Coppa, G.F.; Wang, P. SRT1720, a sirtuin 1 activator, attenuates organ injury and inflammation in sepsis. J. Surg. Res. 2017, 219, 288–295. [Google Scholar] [CrossRef]
- Lemecha, M.; Morino, K.; Imamura, T.; Iwasaki, H.; Ohashi, N.; Ida, S.; Sato, D.; Sekine, O.; Ugi, S.; Maegawa, H. MiR-494-3p regulates mitochondrial biogenesis and thermogenesis through PGC1-α signalling in beige adipocytes. Sci. Rep. 2018, 8, 15096. [Google Scholar] [CrossRef] [Green Version]
- Lima, T.I.; Araujo, H.N.; Menezes, E.S.; Sponton, C.H.; Araújo, M.B.; Bomfim, L.H.; Queiroz, A.L.; Passos, M.A.; Sousa, T.A.E.; Hirabara, S.M. Role of microRNAs on the Regulation of Mitochondrial Biogenesis and Insulin Signaling in Skeletal Muscle. J. Cell. Physiol. 2017, 232, 958–966. [Google Scholar] [CrossRef]
- Ge, Q.M.; Huang, C.M.; Zhu, X.Y.; Bian, F.; Pan, S.M. Differentially expressed miRNAs in sepsis-induced acute kidney injury target oxidative stress and mitochondrial dysfunction pathways. PLoS ONE 2017, 12, e0173292. [Google Scholar] [CrossRef]
- Jesinkey, S.R.; Funk, J.A.; Stallons, L.J.; Wills, L.P.; Megyesi, J.K.; Beeson, C.C.; Schnellmann, R.G. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1157–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baniene, R.; Trumbeckas, D.; Kincius, M.; Pauziene, N.; Raudone, L.; Jievaltas, M.; Trumbeckaite, S. Short ischemia induces rat kidney mitochondria dysfunction. J. Bioenerg. Biomembr. 2016, 48, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Collier, J.B.; Whitaker, R.M.; Eblen, S.T.; Schnellmann, R.G. Rapid Renal Regulation of Peroxisome Proliferator-activated Receptor γ Coactivator-1α by Extracellular Signal-Regulated Kinase 1/2 in Physiological and Pathological Conditions. J. Biol. Chem. 2016, 291, 26850–26859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, A.; Yang, W.L.; Kuncewitch, M.; Jacob, A.; Prince, J.M.; Asirvatham, J.R.; Nicastro, J.; Coppa, G.F.; Wang, P. Sirtuin 1 activation stimulates mitochondrial biogenesis and attenuates renal injury after ischemia-reperfusion. Transplantation 2014, 98, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Lempiäinen, J.; Finckenberg, P.; Mervaala, E.E.; Sankari, S.; Levijoki, J.; Mervaala, E.M. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiol. 2013, 208, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell. Rep. 2019, 27, 1551–1566. [Google Scholar] [CrossRef] [Green Version]
- Li, S.Y.; Park, J.; Qiu, C.; Han, S.H.; Palmer, M.B.; Arany, Z.; Susztak, K. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight 2017, 2, e92930. [Google Scholar] [CrossRef] [Green Version]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J. Cell. Physiol. 2019, 234, 5033–5043. [Google Scholar] [CrossRef]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Koh, S.H.; Shin, S.J.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1α axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Liu, X.; Zhu, X.; Qu, Z.; Gong, Z.; Li, J.; Xiao, L.; Yang, Y.; Liu, H.; Sun, L.; et al. The Role of TLR4 on PGC-1. Oxid. Med. Cell. Longev. 2018, 2018, 6296802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, T.Y.; Park, C.W. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell. Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1α in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 2016, 126, 4205–4218. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Ming, Y.; Xu, C.; Xu, Y.; Zhao, S.; Zhang, Q. Deregulation of long noncoding RNA (TUG1) contributes to excessive podocytes apoptosis by activating endoplasmic reticulum stress in the development of diabetic nephropathy. J. Cell. Physiol. 2019, 234, 15123–15133. [Google Scholar] [CrossRef]
- Yuan, Y.; Huang, S.; Wang, W.; Wang, Y.; Zhang, P.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Activation of peroxisome proliferator-activated receptor-γ coactivator 1α ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012, 82, 771–789. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Yuan, Y.; Bai, M.; Ding, G.; Jia, Z.; Huang, S.; Zhang, A. PGC-1α overexpression protects against aldosterone-induced podocyte depletion: Role of mitochondria. Oncotarget 2016, 7, 12150–12162. [Google Scholar] [CrossRef] [PubMed]
- Aquila, G.; Kostina, A.; Vieceli Dalla Sega, F.; Shlyakhto, E.; Kostareva, A.; Marracino, L.; Ferrari, R.; Rizzo, P.; Malaschicheva, A. The Notch pathway: A novel therapeutic target for cardiovascular diseases? Expert Opin. Ther. Targets 2019, 23, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Ucero, A.C.; Egido, J.; Ruiz-Ortega, M.; Ramos, A.M.; Putterman, C.; Ortiz, A. TWEAK and the progression of renal disease: Clinical translation. Nephrol. Dial. Transplant. 2014, 29 (Suppl. S1), i54–i62. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Dan, L.; Wang, C.; Ma, P.; Yu, Q.; Gu, M.; Dong, L.; Jiang, W.; Pan, S.; Xie, C.; Han, J.; et al. PGC1α promotes cholangiocarcinoma metastasis by upregulating PDHA1 and MPC1 expression to reverse the Warburg effect. Cell. Death Dis. 2018, 9, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, J.H.; Lee, S.H. Hypoxia-induced PGC-1α Regulates Mitochondrial Function and Tumorigenesis of Colorectal Cancer Cells. Anticancer Res. 2019, 39, 4865–4876. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model | Treatment | Effect on PGC-1αand Mitochondria | Effect on Kidney Injury | Reference | |
|---|---|---|---|---|---|
| Folic acid | Anti-TWEAK antibodies | ↑ PGC-1α ↑ MB | ↑ renal function ↓ inflammation | [37] | |
| Crotonate | ↑ PGC-1α | ↑ renal function ↓ inflammation | [145] | ||
| Selective cGMP-specific PDE inhibitors | ↑ PGC-1α ↑ mitochondrial function ↑ MB | ↑ renal function | [34] | ||
| PGC-1α-KO mice | No PGC-1α ↓↓ MB | ↓↓ renal function ↑↑ inflammation ↑↑ tubular cell death | [28] | ||
| NMN | ↑ Sirt1 ↑ mitochondrial function | ↑ renal function | [29] | ||
| Cisplatin | 5-Aminolevulinic acid (even better with Fe) | ↑ PGC-1α ↑ mitochondrial function | ↑ renal function ↓ tubular cell death | [153] | |
| PDK inhibitor DCA / PDK4-KO mice | ↑ PGC-1α ↑ mitochondrial function ↑ MB | ↑ renal function ↓ tubular cell death | [154] | ||
| AICAR / ALCAR | ↑ PGC-1α ↑ mitochondrial function ↓mitochondrial fragmentation, ↓ DRP-1 | ↑ renal function | [155] | ||
| Sepsis | TLR4-KO / Pharmacologic inhibition of MEK/ERK signaling | ↑ PGC-1α ↑ mitochondrial function ↑ MB | ↑ renal function | [162] | |
| STAC (SRT1720) | ↑ Sirt1 | ↑ renal function ↓ inflammation | [167] | ||
| Inducible tubular transgenic mice (iNephPGC1α) | ↑ PGC-1α | ↑ renal function | [31] | ||
| IRI | NAM | ↑ PGC-1α | ↑ renal function ↓ fatty acid accumulation | [31] | |
| Pharmacologic inhibition of MEK/ERK signaling | ↑ PGC-1α ↑ MB | ↑ renal function | [174] | ||
| STAC (SRT1720) | ↑ PGC-1α ↑ mitochondrial function ↑ MB | ↑ renal function ↓ inflammation↓ tubular cell death | [175] | ||
| Caloric restriction | ↑ PGC-1α | ↑ renal function ↓ inflammation | [176] | ||
| Formoterol | Unchanged PGC-1α ↑ mitochondrial function | ↑ renal function | [171] | ||
| PGC-1α-KO mice | Absent PGC-1α | ↓↓ renal function ↓↓ mitochondrial function ↑ fatty acid accumulation | [31] | ||
| Inducible tubular transgenic mice (iNephPGC1α) | ↑ PGC-1α | ↑ renal function ↓ fatty acid accumulation | [31] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. https://doi.org/10.3390/biom10020347
Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, Monsalve M, Ramos AM, Sanchez-Niño MD, Ruiz-Ortega M, Ortiz A, Sanz AB. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules. 2020; 10(2):347. https://doi.org/10.3390/biom10020347
Chicago/Turabian StyleFontecha-Barriuso, Miguel, Diego Martin-Sanchez, Julio Manuel Martinez-Moreno, Maria Monsalve, Adrian Mario Ramos, Maria Dolores Sanchez-Niño, Marta Ruiz-Ortega, Alberto Ortiz, and Ana Belen Sanz. 2020. "The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases" Biomolecules 10, no. 2: 347. https://doi.org/10.3390/biom10020347