Discovery of Kynurenines Containing Oligopeptides as Potent Opioid Receptor Agonists

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. General Procedures
2.2.1. Formation of Ethanolamine-Kynurenic Acid Ester
2.2.2. Coupling Reaction
2.2.3. Amidation
2.2.4. Saponification
2.3. Synthesis and Characterization
2.4. In Vitro Biological Assays
2.4.1. Chemicals
2.4.2. Animal
2.4.3. Preparation of Brain Samples for Binding Assays
2.4.4. Receptor Binding Assays
Functional [35S]GTPγS Binding Experiments
Binding Experiments
Data Analysis
2.5. In Vivo Tests
2.5.1. Animals
2.5.2. Treatment Procedure
2.5.3. Surgery for i.c.v. Injections
2.5.4. Tail Flick Test
2.5.5. Formalin Test
2.5.6. Data Analysis and Statistics
2.6. Stability in Human Plasma Sample Preparation
3. Results and Discussion
3.1. Chemistry
3.2. In Vitro Studies
3.2.1. Binding Assays
3.2.2. Binding-Protein Activation Assays
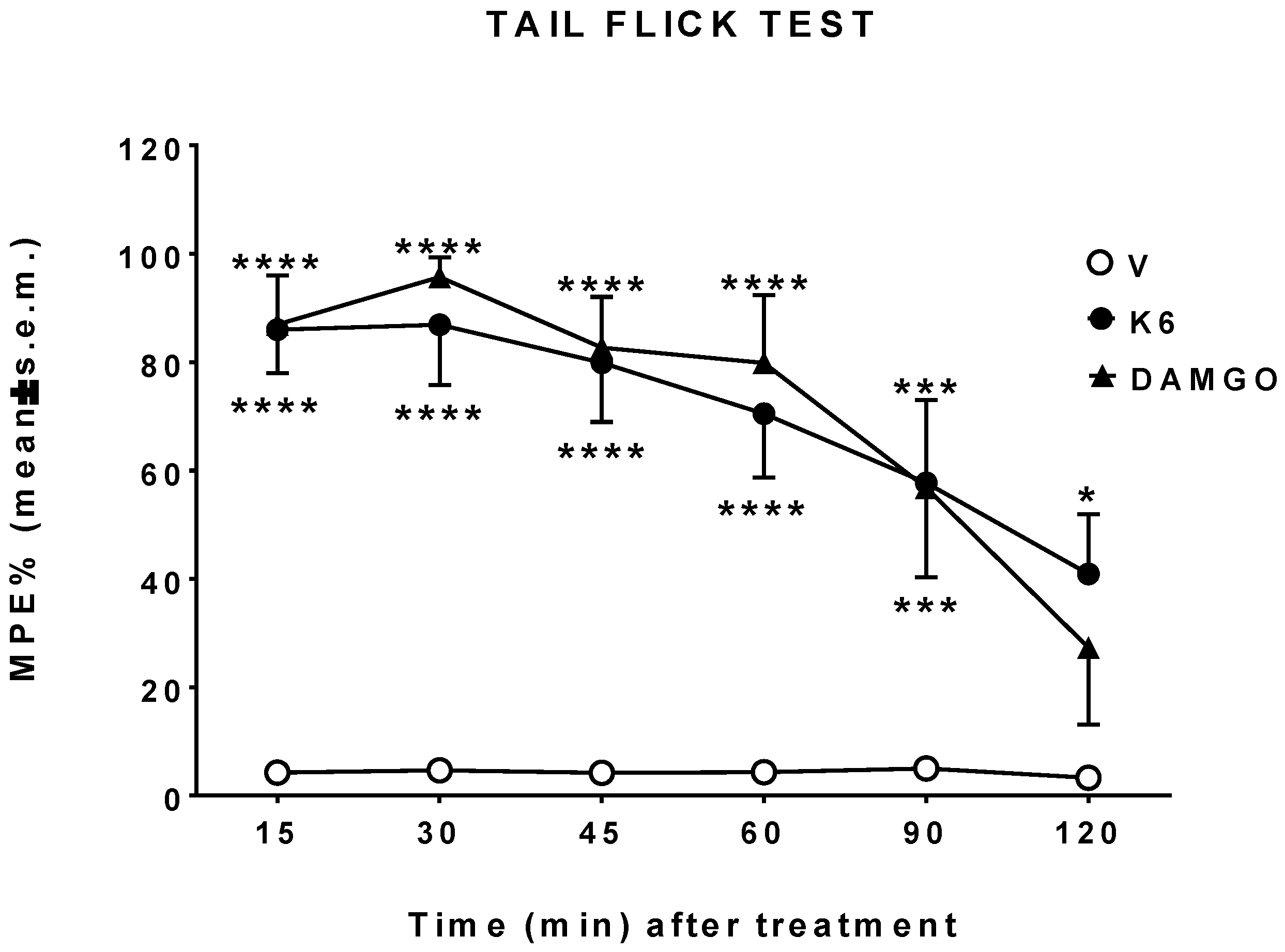
3.3. In Vivo Studies
3.4. Plasma Stability Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| KP | kynurenine pathway |
| kyn | kynurenine |
| kyna | kynurenic acid |
| FDA | Food and Drug Administration |
| CNS | central nervous system |
| BBB | blood brain barrier |
| GPR35 | G protein-coupled receptor 35 |
| SVCT2 | Sodium-dependent vitamin C transporter 2 |
| MOR | μ-opioid receptor; DOR, δ-opioid receptor |
| DOR | δ-opioid receptor |
| KOR | k-opioid receptor; GPCRs, G protein coupled receptors |
| GPCRs | G protein coupled receptors |
| NMR | Nuclear magnetic resonance |
| TFA | trifloroacetic acid |
| ACN | acetonitrile |
| RP-HPLC | Reverse Phase High performance liquid chromatography |
| TMS | trimethylsilane |
| ESI | Electrospray ionization |
| LRMS | Low Resolution Mass Spectroscopy |
| HOBt | 1-hydroxybenzotriazole |
| DMAP | 4-Dimethylaminopyridine |
| EDC.HCl | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| EtOAc | ethyl acetate |
| THF | tetrahydrofurane |
| NMM | N-methylmorpholine |
| iBCF | isobuthylchloroformiate |
| DMSO | dimethylsulfoxide |
| Boc | tert-butyloxycarbonyl |
| Ar | aryl |
| EM-2 | endomorphine-2 |
| BSA | Bovine serum albumin |
| i.c.v. | intracerebroventricular |
| Cyp | cyprodime |
| NTI | naltrindole |
| Nor-BNI | norbinaltorphine |
| DPDPE | [D-Pen,D-Pen5]Enkephalin |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | dimethylformamide |
| DCM | dichloromethane |
| MeOH | methanol |
| TIPS | triisopropylsilane |
| EGTA | ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid |
| GDP | guanosine 5’-diphosphate |
| GTP | guanosine-5’-triphosphate |
| DMSO | dimethylsulfoxide |
| DAMGO | [D-Ala2, N-MePhe4, Gly-ol]-enkephalin |
| IleDelt II | Ile5,6-deltorphin II |
| [35S]GTPγS | guanosine-5’-[35S]thiophosphate |
| NMDA | N-methyl-D-aspartate receptor |
| Tris-HCl | tris-(hydroxymethyl)-aminomethane hydrochloride |
References
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, 9794. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xia, Z.; Zhang, D.; Sheng, Z.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Multifunctional pharmaceutical effects of the antibiotic daptomycin. BioMed Res. Int. 2019, 2019, 8609218. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.-H.; Hsieh, P.-W.; Yang, Y.-L.; Hua, K.-F.; Chang, F.-R.; Shiea, J.; Wu, S.-H.; Wu, Y.-C. Cyclopeptides with Anti-inflammatory Activity from Seeds of Annona Montana. J. Nat. Prod. 2008, 71, 1365–1370. [Google Scholar] [PubMed]
- Ellis-Steinborner, S.T.; Scanlon, D.; Musgrave, I.F.; Nha Tran, T.T.; Hack, S.; Wang, T.; Abell, A.D.; Tyler, M.J.; Bowie, J.H. An unusual kynurenine-containig opioid tetrapeptide from the skin gland secretion of the Australian red tree frog Litoria rubella. Sequence determination by electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug. Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Yeganeh Salehpour, M.; Mollica, A.; Momtaz, S.; Sanadgol, N.; Farzaei, M.H. Melatonin and multiple sclerosis: From plausible neuropharmacological mechanisms of action to experimental and clinical evidence. Clin. Drug Investig. 2019, 39, 607–624. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur. J. Pharmacol. 1988, 151, 313–315. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Actions of kynurenic acid and quinolinic acid in the rat hippocampus in vivo. Exp. Neurol. 1985, 88, 570–579. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a Ligand for Orphan G Protein-coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonina, F.P.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caprariis, P. Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Luhavaya, H.; Sigrist, R.; Chekan, J.R.; McKinnie, S.M.K.; Moore, B.S. Biosynthesis of l-4-Chlorokynurenine, an antidepressant prodrug and a non-proteinogenic amino acid found in lipopeptide. Antibiotics 2019, 58, 8394–8399. [Google Scholar]
- Manfredini, S.; Vertuani, S.; Pavan, B.; Vitali, F.; Scaglianti, M.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.; Dalpiaz, A. Design, synthesis and activity of ascorbic acid prodrugs of nipecotic, kynurenic and diclophenamic acids, liable to increase neurotropic activity. J. Med. Chem. 2002, 45, 559–562. [Google Scholar] [CrossRef]
- Vamos, E.; Pardutz, A.; Klivenyi, P.; Toldi, J.; Vecsei, L. The role of kynurenines in disorders of the central nervous system: Possibilities for neuroprotection. J. Neurol. Sci. 2009, 283, 21–27. [Google Scholar] [CrossRef]
- Knyihár-Csillik, E.; Toldi, J.; Mihály, J.A.; Krisztin-Péva, B.; Chadaide, Z.; Németh, H.; Vécsei, L. Kynurenine in combination with probenecid mitigates the stimulation-induced increase of c-fos immunoreactivity of the rat caudal trigeminal nucleus in an experimental migraine model. J. Neural. Transm. 2007, 114, 417–421. [Google Scholar] [CrossRef]
- Gábor, N.-G.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, A.; Vécsei, L. Interactions between the kynurenine and the endocannabinoid system with special emphasis on migraine. Int. J. Mol. Sci. 2017, 18, 1617. [Google Scholar] [CrossRef]
- Stone, T.W.; Caroline, M.; Forrest, M.; Darlington, L.G. Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J. 2012, 279, 1386–1397. [Google Scholar] [CrossRef]
- Deora, G.S.; Kantham, S.; Chan, S.; Dighe, S.N.; Veliyath, S.K.; McColl, G.; Parat, M.O.; McGeary, R.P.; Ross, B.P. Multifunctional analogs of kynurenic acid for the treatment of Alzheimer’s disease: Synthesis, pharmacology, and molecular modeling studies. ACS Chem. Neurosci. 2017, 8, 2667–2675. [Google Scholar] [CrossRef]
- Zádori, D.; Nyiri, G.; Szonyi, A.; Szatmári, I.; Fülöp, F.; Toldi, J.; Freund, T.F.; Vécsei, L.; Klivényi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neurol. Transm. 2011, 18, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, A.; Pezzoli, D.; Bellucci, M.C.; Malloggi, C.; Negri, A.; Sganappa, A.; Tedeschi, G.; Candiani, G.; Volonterio, A. Synthesis of multifunctional PAMAM-Aminoglycoside conjugates with enhanced transfection efficiency. Bioconjug Chem. 2013, 24, 1928–1936. [Google Scholar] [CrossRef]
- Tsentalovich, Y.P.; Yanshole, V.V.; Polienko, Y.F.; Morozov, S.V.; Grigor’ev, I.A. Deactivation of excited states of kynurenine covalently linked to nitroxides. Photochem. Photobiol. 2011, 87, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Novellino, E.; Mirzaie, S.; Macedonio, G.; Pieretti, S.; Minosi, P.; Szűcs, E.; Erdei, A.I.; Zádor, F.; Benyhe, S.; et al. Opioid receptor activity and analgesic potency of DPDPE peptide analogues containing a xylene bridge. ACS Med. Chem. Lett. 2017, 8, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spetea, M.; Berzetei-Gurske, I.P.; Guerrieri, E.; Schmidhammer, H. Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selectivek-opioid receptor agonist. J. Med. Chem. 2012, 55, 10302–10306. [Google Scholar] [CrossRef] [PubMed]
- Oktem, H.A.; Moitra, J.; Benyhe, S.; Tóth, G.; Lajtha, A.; Borsodi, A. Opioid receptor labeling with the chloromethyl ketone derivative of [3H]Tyr-D-Ala-Gly-(Me)Phe-Gly-ol (DAMGO) II: Covalent labeling of mu opioid binding site by 3H-Tyr-D-Ala-Gly-(Me)Phe chloromethyl ketone. Life Sci. 1991, 48, 1763–1768. [Google Scholar] [CrossRef]
- Guerrieri, E.; Mallareddy, J.R.; Tóth, G.; Schmidhammer, H.; Spetea, M. Synthesis and pharmacological evaluation of [(3)H]HS665, a novel, highly selective radioligand for the kappa opioid receptor. ACS Chem. Neurosci. 2015, 6, 456–463. [Google Scholar] [CrossRef]
- Basu, N.; Scheuhammer, A.M.; Rouvinen-Watt, K.; Grochowina, N.; Evans, R.D.; O’Brien, M.; Chan, H.M. Decreased N-methyl-d-aspartic acid (NMDA) receptor levels are associated with mercury exposure in wild and captive mink. Neurotoxicology 2007, 28, 587–593. [Google Scholar] [CrossRef]
- Benyhe, S.; Farkas, J.; Tóth, G.; Wollemann, M. Met5-enkephalin-Arg6-Phe7, an endogenous neuropeptide, binds to multiple opioid and nonopioid sites in rat brain. J. Neurosci. Res. 1997, 48, 249–258. [Google Scholar] [CrossRef]
- Zádor, F.; Kocsis, D.; Borsodi, A.; Benyhe, S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem. Int. 2014, 67, 14–22. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sim, L.J.; Selley, D.E.; Childers, S.R. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5’-[gamma-[35S]thio]-triphosphate binding. Proc. Natl. Acad. Sci. USA 1995, 92, 7242–7246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynor, J.R.; Nahorski, S.R. Modulation by mu-opioid agonists of guanosine-5’-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [Google Scholar] [PubMed]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Stefanucci, A.; Pinnen, F.; Luisi, G.; Pieretti, S.; Borsodi, A.; Bojinik, E.; Benyhe, S. Hybrid peptides endomorphin-2/DAMGO: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2013, 68, 167–177. [Google Scholar] [CrossRef]
- Stefanucci, A.; Lei, W.; Hruby, V.J.; Macedonio, G.; Luisi, G.; Carradori, S.; Streicher, J.M.; Mollica, A. Fluorescent-labeled bioconjugates of the opioid peptides biphalin and DPDPE incorporating fluorescein-maleimide linkers. Future Med. Chem. 2017, 9, 859–869. [Google Scholar] [CrossRef]
- Zádor, F.; Samavati, R.; Szlavicz, E.; Tuka, B.; Bojnik, E.; Fulop, F.; Toldi, J.; Vecsei, L.; Borsodi, A. Inhibition of opioid receptor mediated G-Protein activity after chronic administration of kynurenic acid and its derivative without direct binding to opioid receptors. CNS Neurol. Disord. Drug Targets 2014, 13, 1520–1529. [Google Scholar] [CrossRef]
- Kemp, J.A.; Grimwood, S.; Foster, A.C. Characterization of the antagonism of excitatory amino acid receptors in rat cortex by kynurenic acid. Br. J. Pharmacol. 1987, 91, 314P. [Google Scholar]
- Cheng, Y.-C.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Márki, A.; Monory, K.; Otvos, F.; Tóth, G.; Krassnig, R.; Schmidhammer, H.; Traynor, J.R.; Roques, B.P.; Maldonado, R.; Borsodi, A. Mu-opioid receptor specific antagonist cyprodime: Characterization by in vitro radioligand and [35S]GTPγS binding assays. Eur. J. Pharmacol. 1999, 383, 209–214. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur. J. Pharmacol. 1988, 146, 185–186. [Google Scholar] [CrossRef]
- Smith, H.S. Peripherally-acting opioids. Pain Physician 2008, 11, 121–132. [Google Scholar]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Shah, K.; Karamyan, V.T.; Abbruscato, T.J. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011, 1383, 307–316. [Google Scholar] [CrossRef]
- Yang, L.; Shah, K.; Wang, H.; Karamyan, V.T.; Abbruscato, T.J. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J. Pharmacol. Exp. Ther. 2011, 339, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Islam, M.R.; Karamyan, V.T.; Abbruscato, T.J. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res. 2015, 1609, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Davis, T.P.; Abbruscato, T.J.; Egleton, R.D. Peptides at the blood brain barrier: Knowing me knowing you. Peptides 2015, 72, 50–56. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Ki + S.E.M. (nM) Opioid System | NMDA System | ||
|---|---|---|---|---|
| DAMGO a | Ile5,6Delt II a | HS665 b | MK-801 a | |
| DAMGO | 0.90 ± 0.28 | n.d. c | n.d. c | n.d. c |
| Ile5,6Delt II | n.d. c | 8.85 ± 0.77 | n.d. c | n.d. c |
| HS665 | n.d. c | n.d. c | 2.38 ± 0.25 | n.d. c |
| MK-801 | n.d. c | n.d. c | n.d. c | 11.45 ± 1.04 |
| EM-2 | 3.16 ± 0.3 | n.d. c | n.d.c | n.d. c |
| KYNA | n.d. c | n.d. c | n.d. c | >10000 |
| L-kyn | n.d. c | n.d. c | n.d. c | >10000 |
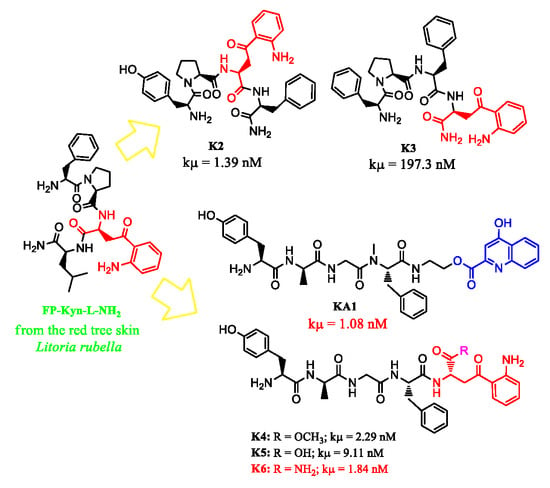
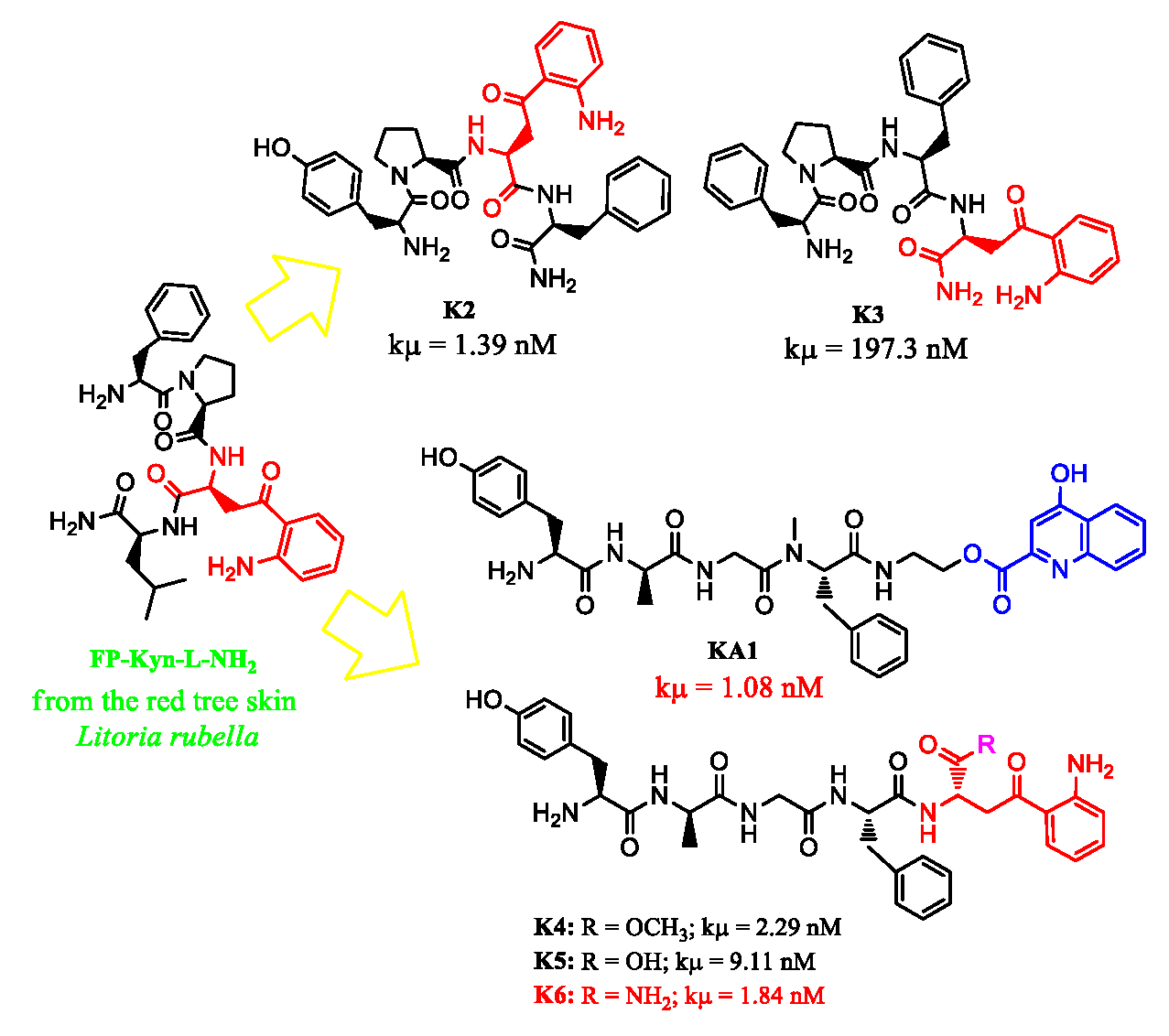
| KA1 | 1.08 ± 0.26 | 554.7 ± 0.8 | >10000 | >10000 |
| K2 | 1.39 ± 0.30 | >10000 | 1043 ± 0.3 | >10000 |
| K3 | 197.3 ± 0.36 | 158.8 ± 1.6 | >10000 | >10000 |
| K4 | 2.29 ± 0.28 | 31.2 ± 0.7 | >10000 | >10000 |
| K5 | 9.11 ± 0.32 | 94.4 ± 0.8 | >10000 | >10000 |
| K6 | 1.84 ± 0.27 | 32.5 ± 0.8 | 127.7 ± 0.3 | >10000 |
| Ligand | Maximal Stimulation (Efficacy) | Potency |
|---|---|---|
| Emax ± S.E.M. (%) | Log EC50 ± S.E.M. | |
| DAMGO | 172.0 ± 3.5 | −6.384 ± 0.101 |
| KA1 | 140.9 ± 1.4 | −6.504 ± 0.076 |
| K2 | 121.6 ± 2.5 | −7.535 ± 0.354 |
| K3 | 114.0 ± 2.1 | −6.993 ± 0.422 |
| K4 | 155.5 ± 4.8 | −6.073 ± 0.172 |
| K5 | 149.2 ± 3.5 | −5.990 ± 0.111 |
| K6 | 209.7 ± 3.4 | −5.984 ± 0.054 |
| Ligand | MOR | DOR | Ligand | KOR | |
|---|---|---|---|---|---|
| Ligand + Cyp | Ligand + NTI | Ligand + Nor-BNI | |||
| Emax + S.E.M. (%) | |||||
| KA1 | 139.6 ± 8.2 | 94.7 ± 3.7 ** | 102.5 ± 1.3 * | 111.7 ± 1.1 | 100.7 ± 6.2 ns |
| K2 | 129.9 ± 8.2 | 88.0 ± 3.3 ** | 88.8 ± 1.9 ** | 92.8 ± 2.0 | 100.7 ± 6.0 ns |
| K3 | 118.9 ± 3.0 | 98.5 ± 1.2 ** | 97.8 ± 1.7 ** | 95.2 ± 2.2 | 106.0 ± 6.3 ns |
| K4 | 160.6 ± 5.7 | 101.6 ± 2.6 *** | 103.6 ± 3.2 *** | 125.2 ± 2.0 | 104.9 ± 9.5 * |
| K5 | 153.1 ± 5.9 | 108.8 ± 1.6 ** | 103.8 ± 1.9 *** | 106.9 ± 0.3 | 110.5 ± 5.0 ns |
| K6 | 211.7 ± 3.1 | 108.5 ± 2.4 *** | 113.3 ± 2.9 *** | 139.8 ± 2.3 | 142.1 ± 7.1 ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szűcs, E.; Stefanucci, A.; Dimmito, M.P.; Zádor, F.; Pieretti, S.; Zengin, G.; Vécsei, L.; Benyhe, S.; Nalli, M.; Mollica, A. Discovery of Kynurenines Containing Oligopeptides as Potent Opioid Receptor Agonists. Biomolecules 2020, 10, 284. https://doi.org/10.3390/biom10020284
Szűcs E, Stefanucci A, Dimmito MP, Zádor F, Pieretti S, Zengin G, Vécsei L, Benyhe S, Nalli M, Mollica A. Discovery of Kynurenines Containing Oligopeptides as Potent Opioid Receptor Agonists. Biomolecules. 2020; 10(2):284. https://doi.org/10.3390/biom10020284
Chicago/Turabian StyleSzűcs, Edina, Azzurra Stefanucci, Marilisa Pia Dimmito, Ferenc Zádor, Stefano Pieretti, Gokhan Zengin, László Vécsei, Sándor Benyhe, Marianna Nalli, and Adriano Mollica. 2020. "Discovery of Kynurenines Containing Oligopeptides as Potent Opioid Receptor Agonists" Biomolecules 10, no. 2: 284. https://doi.org/10.3390/biom10020284