Nrf2—A Molecular Target for Sepsis Patients in Critical Care

 , and
, and
Abstract
:1. Introduction
2. Nrf2—Protection vs. Carcinogenesis
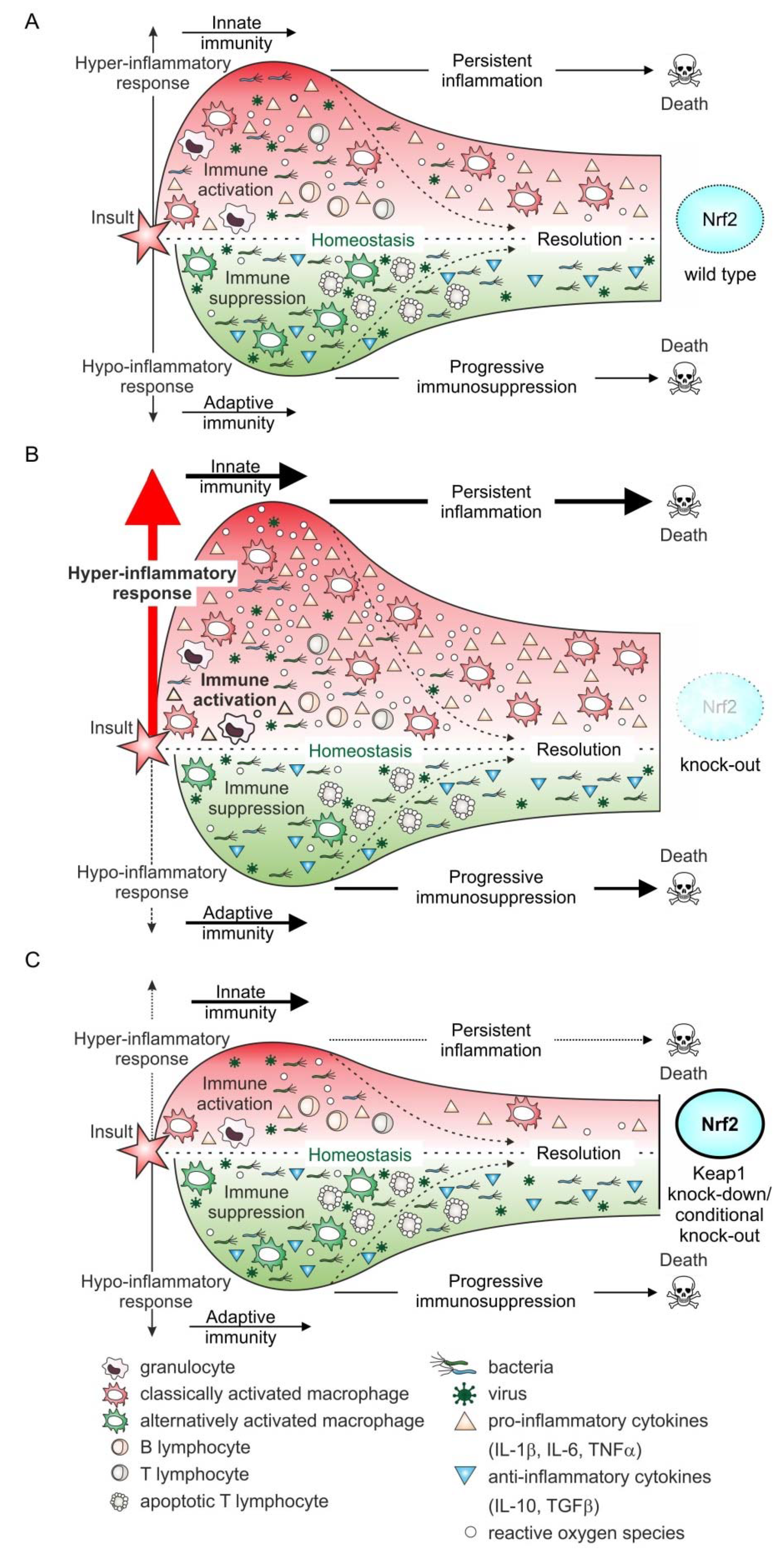
3. Preclinical Sepsis Models of Nrf2 Function
4. Nrf2 in Immune Cell Subpopulations
4.1. B Lymphocytes
4.2. Dendritic Cells (DCs)
4.3. Granulocytes
4.4. Monocytes/Macrophages
4.5. T Lymphocytes
5. Nrf2 in the Sepsis Patient
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, Y.K.; Lee, K.-S.; Cho, D.-H.; Baek, Y.-Y.; Lee, D.-K.; Ha, K.-S.; Choe, J.; Won, M.-H.; Jeoung, D.; et al. Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic. Biol. Med. 2012, 53, 629–640. [Google Scholar] [CrossRef]
- Xu, C.; Li, C.Y.-T.; Kong, A.-N.T. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
- Pall, M.; Levine, S. Nrf2, a master regulator of detoxification and also antioxidant, anti-inflammatory and other cytoprotective mechanisms. Acta Physiol. Sin. 2015, 67, 1–18. [Google Scholar]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D.; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, Y.; Wang, J.; Wang, X.; Chen, C.; Yin, D.; Zhao, F.; Yin, J.; Guo, M.; Zhang, L.; et al. Resveratrol represses estrogen-induced mammary carcinogenesis through NRF2-UGT1A8-estrogen metabolic axis activation. Biochem. Pharmacol. 2018, 155, 252–263. [Google Scholar] [CrossRef]
- Reisman, S.A.; Csanaky, I.L.; Yeager, R.L.; Klaassen, C.D. Nrf2 activation enhances biliary excretion of sulfobromophthalein by inducing glutathione-S-transferase activity. Toxicol. Sci. 2009, 109, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic sulfotransferase as a nephropreventing target by suppression of the uremic toxin indoxyl sulfate accumulation in ischemic acute kidney injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, S.; Mandlekar, S. Role of phase II drug metabolizing enzymes in cancer chemoprevention. Curr. Drug Metab 2009, 10, 595–616. [Google Scholar] [CrossRef]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreger, H.; Westphal, K.; Weller, A.; Baumann, G.; Stangl, V.; Meiners, S.; Stangl, K. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovasc. Res. 2009, 83, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Itoh, K.; Yamamoto, M.; Zweier, J.L.; Li, Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: Protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 2005, 579, 3029–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- DeBlasi, J.M.; DeNicola, G.M. Dissecting the crosstalk between NRF2 signaling and metabolic processes in cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef]
- Martin-Hurtado, A.; Lastres-Becker, I.; Cuadrado, A.; Garcia-Gonzalo, F.R. NRF2 and Primary Cilia: An Emerging Partnership. Antioxidants 2020, 9, 475. [Google Scholar] [CrossRef]
- Liu, P.; Dodson, M.; Fang, D.; Chapman, E.; Zhang, D.D. NRF2 negatively regulates primary ciliogenesis and hedgehog signaling. PLoS Biol. 2020, 18. [Google Scholar] [CrossRef]
- Martin-Hurtado, A.; Martin-Morales, R.; Robledinos-Antón, N.; Blanco, R.; Palacios-Blanco, I.; Lastres-Becker, I.; Cuadrado, A.; Garcia-Gonzalo, F.R. NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling. Sci. Rep. 2019, 9, 13896. [Google Scholar] [CrossRef] [Green Version]
- Reddy, N.M.; Potteti, H.R.; Mariani, T.J.; Biswal, S.; Reddy, S.P. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Thimmulappa, R.; Kombairaju, P.; Biswal, S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J. Immunol. 2010, 185, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouabe, H.; Okkenhaug, K. Gene targeting in mice: A review. Methods Mol. Biol. 2013, 1064, 315–336. [Google Scholar]
- Matthaei, K.I. Genetically manipulated mice: A powerful tool with unsuspected caveats. J. Physiol. 2007, 582, 481–488. [Google Scholar] [CrossRef]
- Remick, D.G.; Ward, P.A. Evaluation of endotoxin models for the study of sepsis. Shock 2005, 24 (Suppl. S1), 7–11. [Google Scholar] [CrossRef]
- Tomasi, M.L.; Ryoo, M.; Yang, H.; Iglesias Ara, A.; Ko, K.S.; Lu, S.C. Molecular mechanisms of lipopolysaccharide-mediated inhibition of glutathione synthesis in mice. Free Radic. Biol. Med. 2014, 68, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Chai, Y.; Lin, H.; Chen, C.; Zhao, M.; Xiong, W.; Zhuang, J.; Fan, X. Dihydroquercetin Activates AMPK/Nrf2/HO-1 Signaling in Macrophages and Attenuates Inflammation in LPS-Induced Endotoxemic Mice. Front. Pharmacol. 2020, 11, 662. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Lai, J.; Yang, L.; Ruan, G.; Chaugai, S.; Ning, Q.; Chen, C.; Wang, D.W. Trimetazidine prevents macrophage-mediated septic myocardial dysfunction via activation of the histone deacetylase sirtuin 1. Br. J. Pharmacol. 2016, 173, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, N.P.; Park, J.-Y.; Lee, H.J.; Kim, S.-S.; Kwon, Y.-S.; Chun, W. Apocynin protects mesangial cells from lipopolysaccharide-induced inflammation by exerting heme oxygenase 1-mediated monocyte chemoattractant protein-1 suppression. Int. J. Mol. Med. 2017, 40, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.; Lang, F.; Chen, Y.; Zhang, H.; Cong, X.; Shen, X.; Su, G. Resveratrol alleviates endotoxin-induced myocardial toxicity via the Nrf2 transcription factor. PLoS ONE 2013, 8, e69452. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, H.J.; Chang, K.C. Glycyrrhizin reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and endotoxemic mice by p38/Nrf2-dependent induction of HO-1. Int. Immunopharmacol. 2015, 26, 112–118. [Google Scholar] [CrossRef]
- Lee, J.-W.; Bae, C.J.; Choi, Y.-J.; Kim, S.-I.; Kwon, Y.-S.; Lee, H.J.; Kim, S.-S.; Chun, W. 3,4,5-trihydroxycinnamic acid inhibits lipopolysaccharide (LPS)-induced inflammation by Nrf2 activation in vitro and improves survival of mice in LPS-induced endotoxemia model in vivo. Mol. Cell. Biochem. 2014, 390, 143–153. [Google Scholar] [CrossRef]
- Planson, A.-G.; Palais, G.; Abbas, K.; Gerard, M.; Couvelard, L.; Delaunay, A.; Baulande, S.; Drapier, J.-C.; Toledano, M.B. Sulfiredoxin protects mice from lipopolysaccharide-induced endotoxic shock. Antioxid. Redox Signal. 2011, 14, 2071–2080. [Google Scholar] [CrossRef]
- Zhong, W.; Qian, K.; Xiong, J.; Ma, K.; Wang, A.; Zou, Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failure by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-κB related signaling. Biomed. Pharmacother. 2016, 83, 302–313. [Google Scholar] [CrossRef]
- Xu, H.; Qi, Q.; Yan, X. Myricetin ameliorates sepsis-associated acute lung injury in a murine sepsis model. Naunyn Schmiedebergs Arch. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Chen, J.; Wang, B.; Lai, J.; Braunstein, Z.; He, M.; Ruan, G.; Yin, Z.; Wang, J.; Cianflone, K.; Ning, Q.; et al. Trimetazidine Attenuates Cardiac Dysfunction in Endotoxemia and Sepsis by Promoting Neutrophil Migration. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, W.J.; Choudhry, M.; Schwacha, M.G.; Kerby, J.D.; Rue, L.W.; Bland, K.I.; Chaudry, I.H. Cecal ligation and puncture. Shock 2005, 24 (Suppl. S1), 52–57. [Google Scholar] [CrossRef]
- Thimmulappa, R.K. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yang, D.; Liu, K.; Hou, L.; Zhang, W. Systematic review and meta-analysis of the protective effect of resveratrol on multiple organ injury induced by sepsis in animal models. Biomed. Rep. 2019, 10, 55–62. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Peng, X.; Zhu, J.; Chen, X.; Liu, H.; Tang, C.; Dong, Z.; Liu, F.; Peng, Y. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am. J. Nephrol. 2014, 40, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Jin, S.; Fei, D.; Kang, K.; Jiang, L.; Lian, Z.; Pan, S.; Zhao, M.; Zhao, M. Artesunate protects against sepsis-induced lung injury via heme oxygenase-1 modulation. Inflammation 2016, 39, 651–662. [Google Scholar] [CrossRef]
- Zong, Y.; Zhang, H. Amentoflavone prevents sepsis-associated acute lung injury through Nrf2-GCLc-mediated upregulation of glutathione. Acta Biochim. Pol. 2017, 64, 93–98. [Google Scholar] [CrossRef]
- Della Giustina, A.; Bonfante, S.; Zarbato, G.F.; Danielski, L.G.; Mathias, K.; de Oliveira, A.N.; Garbossa, L.; Cardoso, T.; Fileti, M.E.; de Carli, R.J.; et al. Dimethyl fumarate modulates oxidative stress and inflammation in organs after sepsis in rats. Inflammation 2018, 41, 315–327. [Google Scholar] [CrossRef]
- Zarbato, G.F.; de Souza Goldim, M.P.; Della Giustina, A.; Danielski, L.G.; Mathias, K.; Florentino, D.; de Oliveira Junior, A.N.; Da Rosa, N.; Laurentino, A.O.; Trombetta, T.; et al. Dimethyl fumarate limits neuroinflammation and oxidative stress and improves cognitive impairment after polymicrobial sepsis. Neurotox. Res. 2018, 34, 418–430. [Google Scholar] [CrossRef]
- Kim, S.R.; Ha, Y.M.; Kim, Y.M.; Park, E.J.; Kim, J.W.; Park, S.W.; Kim, H.J.; Chung, H.T.; Chang, K.C. Ascorbic acid reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and improves survival rate in septic mice by activation of Nrf2/HO-1 signals. Biochem. Pharmacol. 2015, 95, 279–289. [Google Scholar] [CrossRef]
- Doi, K. Role of kidney injury in sepsis. J. Intensive Care 2016, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Mertens, C.; Kuchler, L.; Sola, A.; Guiteras, R.; Grein, S.; Brüne, B.; von Knethen, A.; Jung, M. Macrophage-derived iron-bound lipocalin-2 correlates with renal recovery markers following sepsis-induced kidney damage. Int. J. Mol. Sci. 2020, 21, 7527. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Lin, H.; Liu, H.; Lu, Z.; Wang, H.; Fan, S.; Li, N. Honokiol attenuates sepsis-associated acute kidney injury via the inhibition of oxidative stress and inflammation. Inflammation 2019, 42, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-Y.; Sheng, Z.-X.; Yao, H. Pachymic acid ameliorates sepsis-induced acute kidney injury by suppressing inflammation and activating the Nrf2/HO-1 pathway in rats. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1924–1931. [Google Scholar]
- Wang, Y.; Feng, F.; Liu, M.; Xue, J.; Huang, H. Resveratrol ameliorates sepsis-induced acute kidney injury in a pediatric rat model via Nrf2 signaling pathway. Exp. Ther. Med. 2018, 16, 3233–3240. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.-K.; Hu, L.-L.; Zhang, Y.-X.; Xu, Y.-L.; Liu, X.-Y.; He, P.-K.; Jia, Y.-H. 6-Gingerol ameliorates sepsis-induced liver injury through the Nrf2 pathway. Int. Immunopharmacol. 2020, 80, 106196. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chhibber, S. Intravenous 2-chloroadenosine protects BALB/c mice from Klebsiella pneumoniae B5055-induced sepsis by modulating the pro-inflammatory immune response. J. Chemother. 2009, 21, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Toky, V.; Sharma, S.; Arora, B.B.; Chhibber, S. Establishment of a sepsis model following implantation of Klebsiella pneumoniae-infected fibrin clot into the peritoneal cavity of mice. Folia Microbiol. 2003, 48, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wochal, P.; Rathinam, V.A.K.; Dunne, A.; Carlson, T.; Kuang, W.; Seidl, K.J.; Hall, J.P.; Lin, L.-L.; Collins, M.; Schattgen, S.A.; et al. TRIL is involved in cytokine production in the brain following Escherichia coli infection. J. Immunol. 2014, 193, 1911–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.R.; Clabots, C.; Hirt, H.; Waters, C.; Dunny, G. Enterococcal aggregation substance and binding substance are not major contributors to urinary tract colonization by Enterococcus faecalis in a mouse model of ascending unobstructed urinary tract infection. Infect. Immun. 2004, 72, 2445–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Stirling, B.; Derry, J.; Koga, T.; Jono, H.; Woo, C.-H.; Xu, H.; Bourne, P.; Ha, U.-H.; Ishinaga, H.; et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity 2007, 27, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Chhibber, S. Acute lung inflammation in Klebsiella pneumoniae B5055-induced pneumonia and sepsis in BALB/c mice: A comparative study. Inflammation 2011, 34, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Traeger, T.; Entleutner, M.; Westerholt, A.; Kleist, B.; Hüser, N.; Holzmann, B.; Stier, A.; Pfeffer, K.; Heidecke, C.-D. Cecal ligation and puncture versus colon ascendens stent peritonitis: Two distinct animal models for polymicrobial sepsis. Shock 2004, 21, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Zantl, N.; Uebe, A.; Neumann, B.; Wagner, H.; Siewert, J.R.; Holzmann, B.; Heidecke, C.D.; Pfeffer, K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect. Immun. 1998, 66, 2300–2309. [Google Scholar] [CrossRef] [Green Version]
- Starr, M.E.; Steele, A.M.; Saito, M.; Hacker, B.J.; Evers, B.M.; Saito, H. A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS ONE 2014, 9, e115705. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Platt, M.P.; Fu, H.; Gui, Y.; Wang, Y.; Gonzalez-Juarbe, N.; Zhou, D.; Yu, Y. Global proteome and phosphoproteome characterization of sepsis-induced kidney injury. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef]
- von Knethen, A.; Schäfer, A.; Kuchler, L.; Knape, T.; Christen, U.; Hintermann, E.; Fißlthaler, B.; Schröder, K.; Brandes, R.P.; Genz, B.; et al. Tolerizing CTL by sustained hepatic PD-L1 expression provides a new therapy approach in mouse sepsis. Theranostics 2019, 9, 2003–2016. [Google Scholar] [CrossRef]
- Leeds, J.; Scindia, Y.; Loi, V.; Wlazlo, E.; Ghias, E.; Cechova, S.; Portilla, D.; Ledesma, J.; Swaminathan, S. Protective role of DJ-1 in endotoxin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2020. [Google Scholar] [CrossRef]
- Ehrentraut, H.; Meyer, R.; Schwederski, M.; Ehrentraut, S.; Velten, M.; Grohé, C.; Knuefermann, P.; Baumgarten, G.; Boehm, O. Systemically administered ligands of Toll-like receptor 2, -4, and -9 induce distinct inflammatory responses in the murine lung. Mediat. Inflamm. 2011, 2011, 746532. [Google Scholar] [CrossRef] [PubMed]
- Checconi, P.; de Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-modulating agents in the treatment of viral infections. Int. J. Mol. Sci. 2020, 21, 4084. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K. Oxidative stress in sepsis: A redox redux. J. Clin. Investig. 2006, 116, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Yates, M.S.; Dolan, P.D.; Chen, S.; Liby, K.T.; Sporn, M.B.; Taguchi, K.; Yamamoto, M.; Kensler, T.W. Genetic or pharmacologic amplification of Nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol. Sci. 2008, 104, 218–227. [Google Scholar] [CrossRef]
- Kim, S.-J.; Habib, O.; Kim, J.-S.; Han, H.-W.; Koo, S.K.; Kim, J.-H. A homozygous Keap1-knockout human embryonic stem cell line generated using CRISPR/Cas9 mediates gene targeting. Stem Cell Res. 2017, 19, 52–54. [Google Scholar] [CrossRef]
- Noel, S.; Lee, S.A.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. KEAP1 editing using CRISPR/Cas9 for therapeutic NRF2 activation in pimary human T lymphocytes. J. Immunol. 2018, 200, 1929–1936. [Google Scholar]
- Cho, H.-Y.; Marzec, J.; Kleeberger, S.R. Functional polymorphisms in Nrf2: Implications for human disease. Free Radic. Biol. Med. 2015, 88, 362–372. [Google Scholar] [CrossRef]
- Marzec, J.M.; Christie, J.D.; Reddy, S.P.; Jedlicka, A.E.; Vuong, H.; Lanken, P.N.; Aplenc, R.; Yamamoto, T.; Yamamoto, M.; Cho, H.-Y.; et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007, 21, 2237–2246. [Google Scholar] [CrossRef]
- Madden, S.K.; Itzhaki, L.S. Structural and mechanistic insights into the Keap1-Nrf2 system as a route to drug discovery. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140405. [Google Scholar] [CrossRef]
- Schmoll, D.; Engel, C.K.; Glombik, H. The Keap1-Nrf2 protein-protein interaction: A suitable target for small molecules. Drug Discov. Today Technol. 2017, 24, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Yim, S.H.; Garcia-Manteiga, J.M.; Masciarelli, S.; Kim, Y.-J.; Kang, M.-H.; Iuchi, Y.; Fujii, J.; Vené, R.; Rubartelli, A.; et al. B- to plasma-cell terminal differentiation entails oxidative stress and profound reshaping of the antioxidant responses. Antioxid. Redox Signal. 2010, 13, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Bursley, J.K.; Rockwell, C.E. Nrf2-dependent and -independent effects of tBHQ in activated murine B cells. Food Chem. Toxicol. 2020, 145, 111595. [Google Scholar] [CrossRef] [PubMed]
- Bancos, S.; Baglole, C.J.; Rahman, I.; Phipps, R.P. Induction of heme oxygenase-1 in normal and malignant B lymphocytes by 15-deoxy-Delta(12,14)-prostaglandin J(2) requires Nrf2. Cell. Immunol. 2010, 262, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Aw Yeang, H.X.; Hamdam, J.M.; Al-Huseini, L.M.A.; Sethu, S.; Djouhri, L.; Walsh, J.; Kitteringham, N.; Park, B.K.; Goldring, C.E.; Sathish, J.G. Loss of transcription factor nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) leads to dysregulation of immune functions, redox homeostasis, and intracellular signaling in dendritic cells. J. Biol. Chem. 2012, 287, 10556–10564. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef] [Green Version]
- Helou, D.G.; Braham, S.; de Chaisemartin, L.; Granger, V.; Damien, M.-H.; Pallardy, M.; Kerdine-Römer, S.; Chollet-Martin, S. Nrf2 downregulates zymosan-induced neutrophil activation and modulates migration. PLoS ONE 2019, 14, e0216465. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Werner, S. Nrf2 is highly expressed in neutrophils, but myeloid cell-derived Nrf2 is dispensable for wound healing in mice. PLoS ONE 2017, 12, e0187162. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Fuchs, R.J.; Malhotra, D.; Scollick, C.; Traore, K.; Bream, J.H.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid. Redox Signal. 2007, 9, 1963–1970. [Google Scholar] [CrossRef]
- Furuya, A.K.M.; Sharifi, H.J.; Jellinger, R.M.; Cristofano, P.; Shi, B.; de Noronha, C.M.C. Sulforaphane inhibits HIV infection of macrophages through Nrf2. PLoS Path. 2016, 12, e1005581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, L.K.; Sha, W.; Kuchler, L.; Daiber, A.; Giegerich, A.K.; Weigert, A.; Knape, T.; Snodgrass, R.; Schröder, K.; Brandes, R.P.; et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8(+) T cell function by limiting GSH and Cys availability. Free Radic. Biol. Med. 2015, 83, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Pyaram, K.; Kumar, A.; Kim, Y.-H.; Noel, S.; Reddy, S.P.; Rabb, H.; Chang, C.-H. Keap1-Nrf2 system plays an important role in invariant natural killer T cell development and homeostasis. Cell. Rep. 2019, 27, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Klemm, P.; Rajendiran, A.; Fragoulis, A.; Wruck, C.; Schippers, A.; Wagner, N.; Bopp, T.; Tenbrock, K.; Ohl, K. Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur. J. Immunol. 2020, 50, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Su, W.; Wan, T.; Yu, J.; Zhu, W.; Tang, F.; Liu, G.; Olsen, N.; Liang, D.; Zheng, S.G. Sodium butyrate regulates Th17/Treg cell balance to ameliorate uveitis via the Nrf2/HO-1 pathway. Biochem. Pharmacol. 2017, 142, 111–119. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; AlThagfan, S.S.; Alqinyah, M.; Alqahtani, F.; Ibrahim, K.E.; Al-Harbi, M.M. Sulforaphane treatment reverses corticosteroid resistance in a mixed granulocytic mouse model of asthma by upregulation of antioxidants and attenuation of Th17 immune responses in the airways. Eur. J. Pharmacol. 2019, 855, 276–284. [Google Scholar] [CrossRef]
- Tsai, J.J.; Velardi, E.; Shono, Y.; Argyropoulos, K.V.; Holland, A.M.; Smith, O.M.; Yim, N.L.; Rao, U.K.; Kreines, F.M.; Lieberman, S.R.; et al. Nrf2 regulates CD4+ T cell-induced acute graft-versus-host disease in mice. Blood 2018, 132, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.A.; Reddy, S.P.; Rabb, H. T lymphocyte-specific activation of Nrf2 protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagorski, J.W.; Turley, A.E.; Freeborn, R.A.; VanDenBerg, K.R.; Dover, H.E.; Kardell, B.R.; Liby, K.T.; Rockwell, C.E. Differential effects of the Nrf2 activators tBHQ and CDDO-Im on the early events of T cell activation. Biochem. Pharmacol. 2018, 147, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morzadec, C.; Macoch, M.; Sparfel, L.; Kerdine-Römer, S.; Fardel, O.; Vernhet, L. Nrf2 expression and activity in human T lymphocytes: Stimulation by T cell receptor activation and priming by inorganic arsenic and tert-butylhydroquinone. Free Radic. Biol. Med. 2014, 71, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- Bertolotti, M.; Sitia, R.; Rubartelli, A. On the redox control of B lymphocyte differentiation and function. Antioxid. Redox Signal. 2012, 16, 1139–1149. [Google Scholar] [CrossRef]
- Abis, G.; Charles, R.L.; Kopec, J.; Yue, W.W.; Atkinson, R.A.; Bui, T.T.T.; Lynham, S.; Popova, S.; Sun, Y.-B.; Fraternali, F.; et al. 15-deoxy-Δ12,14-Prostaglandin J2 inhibits human soluble epoxide hydrolase by a dual orthosteric and allosteric mechanism. Commun. Biol. 2019, 2, 188. [Google Scholar] [CrossRef]
- Piesche, M.; Roos, J.; Kühn, B.; Fettel, J.; Hellmuth, N.; Brat, C.; Maucher, I.V.; Awad, O.; Matrone, C.; Comerma Steffensen, S.G.; et al. The emerging therapeutic potential of nitro fatty acids and other Michael acceptor-containing drugs for the treatment of inflammation and cancer. Front. Pharmacol. 2020, 11, 1297. [Google Scholar] [CrossRef]
- Maucher, I.V.; Rühl, M.; Kretschmer, S.B.M.; Hofmann, B.; Kühn, B.; Fettel, J.; Vogel, A.; Flügel, K.T.; Manolikakes, G.; Hellmuth, N.; et al. Michael acceptor containing drugs are a novel class of 5-lipoxygenase inhibitor targeting the surface cysteines C416 and C418. Biochem. Pharmacol. 2017, 125, 55–74. [Google Scholar] [CrossRef]
- Awwad, K.; Steinbrink, S.D.; Frömel, T.; Lill, N.; Isaak, J.; Häfner, A.-K.; Roos, J.; Hofmann, B.; Heide, H.; Geisslinger, G.; et al. Electrophilic fatty acid species inhibit 5-lipoxygenase and attenuate sepsis-induced pulmonary inflammation. Antioxid. Redox Signal. 2014, 20, 2667–2680. [Google Scholar] [CrossRef] [Green Version]
- Al-Huseini, L.M.A.; Aw Yeang, H.X.; Sethu, S.; Alhumeed, N.; Hamdam, J.M.; Tingle, Y.; Djouhri, L.; Kitteringham, N.; Park, B.K.; Goldring, C.E.; et al. Nuclear factor-erythroid 2 (NF-E2) p45-related factor-2 (Nrf2) modulates dendritic cell immune function through regulation of p38 MAPK-cAMP-responsive element binding protein/activating transcription factor 1 signaling. J. Biol. Chem. 2013, 288, 22281–22288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Bittner, C.; Leyck, S. Changes in glutathione peroxidase activities and the oxidative burst of leukocytes during inflammation in the mouse and rat. Free Radic. Res. Commun. 1987, 4, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Culić, O.; Eraković, V.; Munić, V.; Popović-Grle, S.; Barisić, K.; Bosnar, M.; Brajsa, K.; Cepelak, I.; Cuzić, S.; et al. Modulation of neutrophil and inflammation markers in chronic obstructive pulmonary disease by short-term azithromycin treatment. Eur. J. Pharmacol. 2005, 517, 132–143. [Google Scholar] [CrossRef]
- Dominis-Kramarić, M.; Bosnar, M.; Kelnerić, Z.; Glojnarić, I.; Cužić, S.; Parnham, M.J.; Eraković Haber, V. Comparison of pulmonary inflammatory and antioxidant responses to intranasal live and heat-killed Streptococcus pneumoniae in mice. Inflammation 2011, 34, 471–486. [Google Scholar] [CrossRef]
- Riksen, N.P.; Netea, M.G. Immunometabolic control of trained immunity. Mol. Asp. Med. 2020, 100897. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [Green Version]
- Hooftman, A.; O’Neill, L.A.J. The immunomodulatory potential of the metabolite itaconate. Trends Immunol. 2019, 40, 687–698. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Wruck, C.J.; Streetz, K.; Pavic, G.; Götz, M.E.; Tohidnezhad, M.; Brandenburg, L.-O.; Varoga, D.; Eickelberg, O.; Herdegen, T.; Trautwein, C.; et al. Nrf2 induces interleukin-6 (IL-6) expression via an antioxidant response element within the IL-6 promoter. J. Biol. Chem. 2011, 286, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-Y.; Chen, C.; Wang, L.; Chang, X.; Zheng, P.; Liu, Y. Cutting edge: Broad expression of the FoxP3 locus in epithelial cells: A caution against early interpretation of fatal inflammatory diseases following in vivo depletion of FoxP3-expressing cells. J. Immunol. 2008, 180, 5163–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerboni, S.; Gehrmann, U.; Preite, S.; Mitra, S. Cytokine regulated Th17 plasticity in human health and diseases. Immunology 2020. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Vitturi, D.A.; Jorkasky, D.K.; Freeman, B.A. Nitro-fatty acids: New drug candidates for chronic inflammatory and fibrotic diseases. Nitric Oxide 2018, 79, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current landscape of NRF2 biomarkers in clinical trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- Senoglu, N.; Yuzbasioglu, M.F.; Aral, M.; Ezberci, M.; Kurutas, E.B.; Bulbuloglu, E.; Ezberci, F.; Oksuz, H.; Ciragil, P. Protective effects of N-acetylcysteine and beta-glucan pretreatment on oxidative stress in cecal ligation and puncture model of sepsis. J. Investig. Surg. 2008, 21, 237–243. [Google Scholar] [CrossRef]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020, 156, 107–112. [Google Scholar] [CrossRef]
- Noguchi, N. Ebselen, a useful tool for understanding cellular redox biology and a promising drug candidate for use in human diseases. Arch. Biochem. Biophys. 2016, 595, 109–112. [Google Scholar] [CrossRef]
- Parnham, M.J.; Sies, H. The early research and development of ebselen. Biochem. Pharmacol. 2013, 86, 1248–1253. [Google Scholar] [CrossRef]
- Peivandi Yazdi, A.; Razavi, M.; Sheikh, S.; Boroumand, N.; Salehi, M.; Hashemy, S.I. Clinical trial assessment of intermittent and continuous infusion dose of N-acetylcysteine on redox status of the body in patients with sepsis admitted to the ICU. J. Intensive Care Med. 2019, 35, 1383–1388. [Google Scholar] [CrossRef]
- Chertoff, J. N-acetylcysteine’s role in sepsis and potential benefit in patients with microcirculatory derangements. J. Intensive Care Med. 2018, 33, 87–96. [Google Scholar] [CrossRef]
- Najafi, A.; Mojtahedzadeh, M.; Ahmadi, K.H.; Abdollahi, M.; Mousavi, M.; Chelkeba, L.; Najmeddin, F.; Ahmadi, A. The immunological benefit of higher dose N-acetyl cysteine following mechanical ventilation in critically ill patients. Daru 2014, 22, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spapen, H.D.; Diltoer, M.W.; Nguyen, D.N.; Hendrickx, I.; Huyghens, L.P. Effects of N-acetylcysteine on microalbuminuria and organ failure in acute severe sepsis: Results of a pilot study. Chest 2005, 127, 1413–1419. [Google Scholar] [CrossRef]
- Montull, B.; Menéndez, R.; Torres, A.; Méndez, R. Predictors of severe sepsis among patients hospitalized for community-acquired pneumonia. MLO Med. Lab. Obs. 2016, 48, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, A.R.; Groth, G.; Heller, S.C.; Breitkreutz, R.; Nebe, T.; Quintel, M.; Koch, T. N-acetylcysteine reduces respiratory burst but augments neutrophil phagocytosis in intensive care unit patients. Crit. Care Med. 2001, 29, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jiménez-Villegas, J.; Escoll, M.; Fernández-Ginés, R.; Garcia Yagüe, A.J.; Lastra, D.; Manda, G.; Rojo, A.I.; et al. Can Activation of NRF2 Be a Strategy against COVID-19? Trends Pharmacol. Sci. 2020, 41, 598–610. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune Cell Subtype | Model | Role of Nrf2 Expression/Activation | Ref. |
|---|---|---|---|
| B lymphocytes | terminal plasma cell differentiation | Nrf2 target gene expression↑, plasma cell differentiation↓↑ | [102] |
| Nrf2 WT vs. KO mice, splenocytes-derived B cells, LPS-treated, tBHQ | IgM↑, CD25↓, CD69↓, CD22↓, plasma cell differentiation (CD138) ↓ | [103] | |
| PBMC derived B cells, 15-dPGJ2 | HO-1↑ | [104] | |
| Dendritic cells | PBMC-derived DCs, glioma-dependent microenvironment Nrf2 siRNA | DC maturation and activation↓, restored by Nrf2 knockdown | [105] |
| Nrf2 WT vs. knock-out mice, BM-derived DCs | iDC: GSH↑, CD80/CD86↓, phagocytosis↑, antigen presentation↑ | [106] | |
| Nrf2 WT vs. knock-out mice, BM- and myeloid lung derived DCs | pro-inflammatory markers↓ | [107] | |
| Granulocytes | siNrf2, TMZ | migration↑ | [59] |
| Nrf2 WT vs. KO mice, | zymosan-dependent activation↑ and migration↑ | [108] | |
| primary neutrophils | Nrf2 is highly expressed | [109] | |
| primary neutrophils (PBMCs), LPS treatment RTA-402 | pro-inflammatory gene expression↓ | [110] | |
| Monocytes/ Macrophages | HIV, SFN | HIV infection↓ | [111] |
| Nrf2 WT vs. KO mice BM-derived MΦ | antigen-driven CD8+ T cell function↓ by limiting MΦ -dependent GSH and Cys availability | [112] | |
| endotoxin model [15 mg/kg], LPS treated primary human MΦ derived from PBMCs and primary mouse MΦ BM-derived | itaconate-dependent activation of Nrf2 linking Nrf2 activation to cell metabolism | [113] | |
| COPD, intranasal application of H. influenza and P. aeruginosa, alveolar MΦ; SFN | MARCO expression↑, bacterial phagocytosis↑ | [114] | |
| BM-derived myeloid lineage specific Keap1 KO (LysMCre/Keap1fl/fl) MΦ, M1 vs. M2 polarized, DEM, 15d-PGJ2 | MΦ M1-induced gene expression ↓ MΦ M2-induced gene expression↓↑ | [115] | |
| T lymphocytes | invariant natural killer T cells, CD4+ T cell specific Keap1 knock-out mice or Nrf2 knock-out | NKT cell development, homeostasis, and metabolism↓, NKT2 and NKT17↑, NKT1↓, additional Nrf2 knock-out restored all Keap1 mediated effects | [116] |
| Tregs, Foxp3-specific Keap1 KO | Tregs↓ | [117] | |
| Nrf2 WT mice, experimental autoimmune uveitis, draining lymph nodes, spleen, NaB | Th17↓, Tregs↑, HO-1↑ | [118] | |
| Th17, mixed granulocyte airway inflammation, sensitization and challenge via cockroach allergen extract, SFN | Th17↓, IL-6↓, IL-17A↓, IL-23↓ | [119] | |
| Nrf2 WT vs. KO mice, CD4+ and CD8+ T cells, acute graft versus host disease in mice | CD4+ T cells↓, CD8+ T cells↓↑, nTreg Helios+↑ | [120] | |
| CRISPR/Cas-dependent Keap1 knock down in Jurkat T cells and primary human T lymphocytes | CD4+ T cells↑, CD8+ T cells↓, Nrf2-dependent target genes↑ | [97] | |
| CD4+ T cell specific Keap1 knock-out mice, IR-induced AKI | baseline antioxidant gene expression↑, intrarenal CD25+Foxp3+ Tregs↑, intracellular level of TNFα, IFNγ, and IL-17↓, AKI↓ | [121] | |
| Nrf2 WT vs. knock-out mice, splenocytes, tBHQ, CDDO-Im | TNFα↓, IFNγ↓, IL-2↑, CD25↓↑, CD69↓↑, NF-κB↓, c-Jun↑, Th1↓, Th2↑ | [122] | |
| Nrf2 WT vs. knock-out mice, lymph nodes, spleen, αCD3/CD28-ab treated, tBHQ | IFNγ↓, Nrf2 target genes (NQO1, GCLC, HO-1); Th2 markers (IL-4, IL-5, IL-13)↑ | [123] | |
| human CD4+ T cells derived from PBMCs, αCD3/CD28-ab treated, Nrf2 siRNA, tBHQ | Nrf2 target genes (NQO1, GCLM, HO-1, TRX1)↑, activation markers (IL-2, TNFα, IFNγ, IL17)↓↑ | [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunne, S.; Heinicke, U.; Parnham, M.J.; Laux, V.; Zacharowski, K.; von Knethen, A. Nrf2—A Molecular Target for Sepsis Patients in Critical Care. Biomolecules 2020, 10, 1688. https://doi.org/10.3390/biom10121688
Gunne S, Heinicke U, Parnham MJ, Laux V, Zacharowski K, von Knethen A. Nrf2—A Molecular Target for Sepsis Patients in Critical Care. Biomolecules. 2020; 10(12):1688. https://doi.org/10.3390/biom10121688
Chicago/Turabian StyleGunne, Sandra, Ulrike Heinicke, Michael J. Parnham, Volker Laux, Kai Zacharowski, and Andreas von Knethen. 2020. "Nrf2—A Molecular Target for Sepsis Patients in Critical Care" Biomolecules 10, no. 12: 1688. https://doi.org/10.3390/biom10121688