Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fresh Frozen Clinical Samples
2.2. Cell Lines and Cell Culture Conditions
2.3. Small Interfering RNA (siRNA) Transfection
2.4. RNA Extraction and Quantitative Real-Time Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
2.5. Cell Viability Assay
2.6. Cell Cycle Analysis
2.7. Detection of Apoptosis
2.8. Immunohistochemical Staining (IHC)
2.9. Protein Extraction and Western Blotting
2.10. Colony Formation Assay
2.11. Statistical Analyses
3. Results
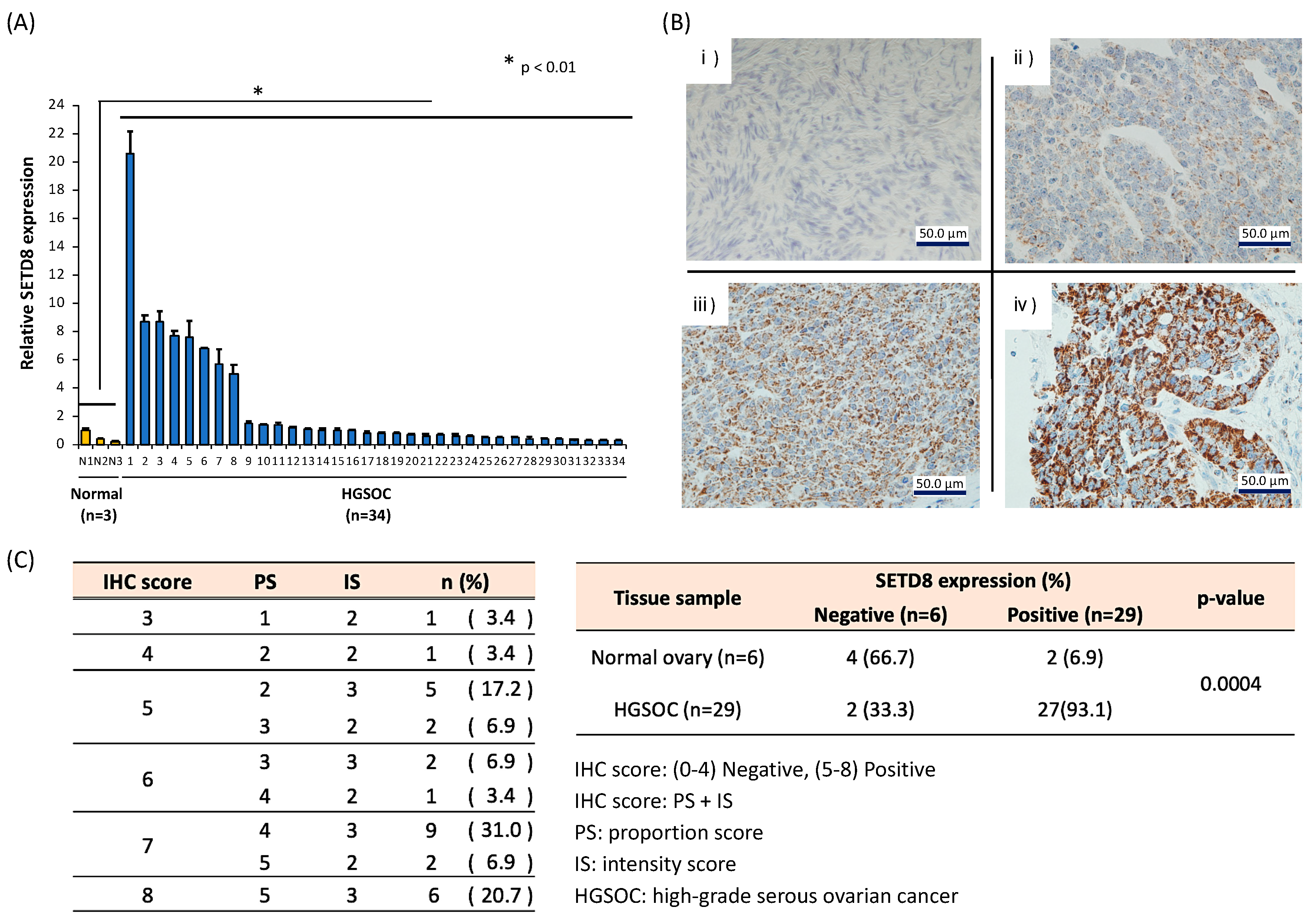
3.1. Expression Profiling of Histone Methyltransferases Identifies SETD8 as Overexpressed in HGSOC Cell Lines and Tissues
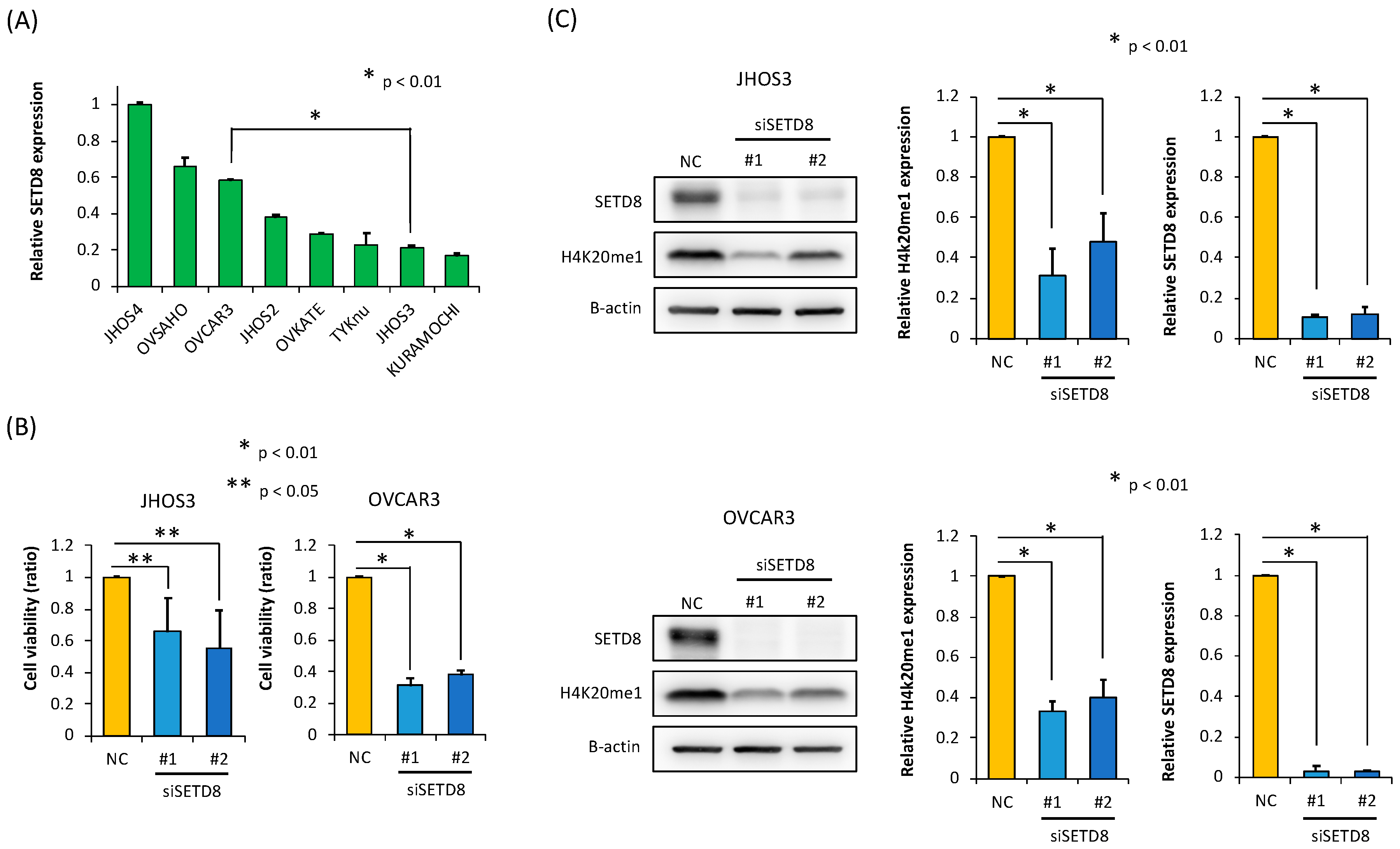
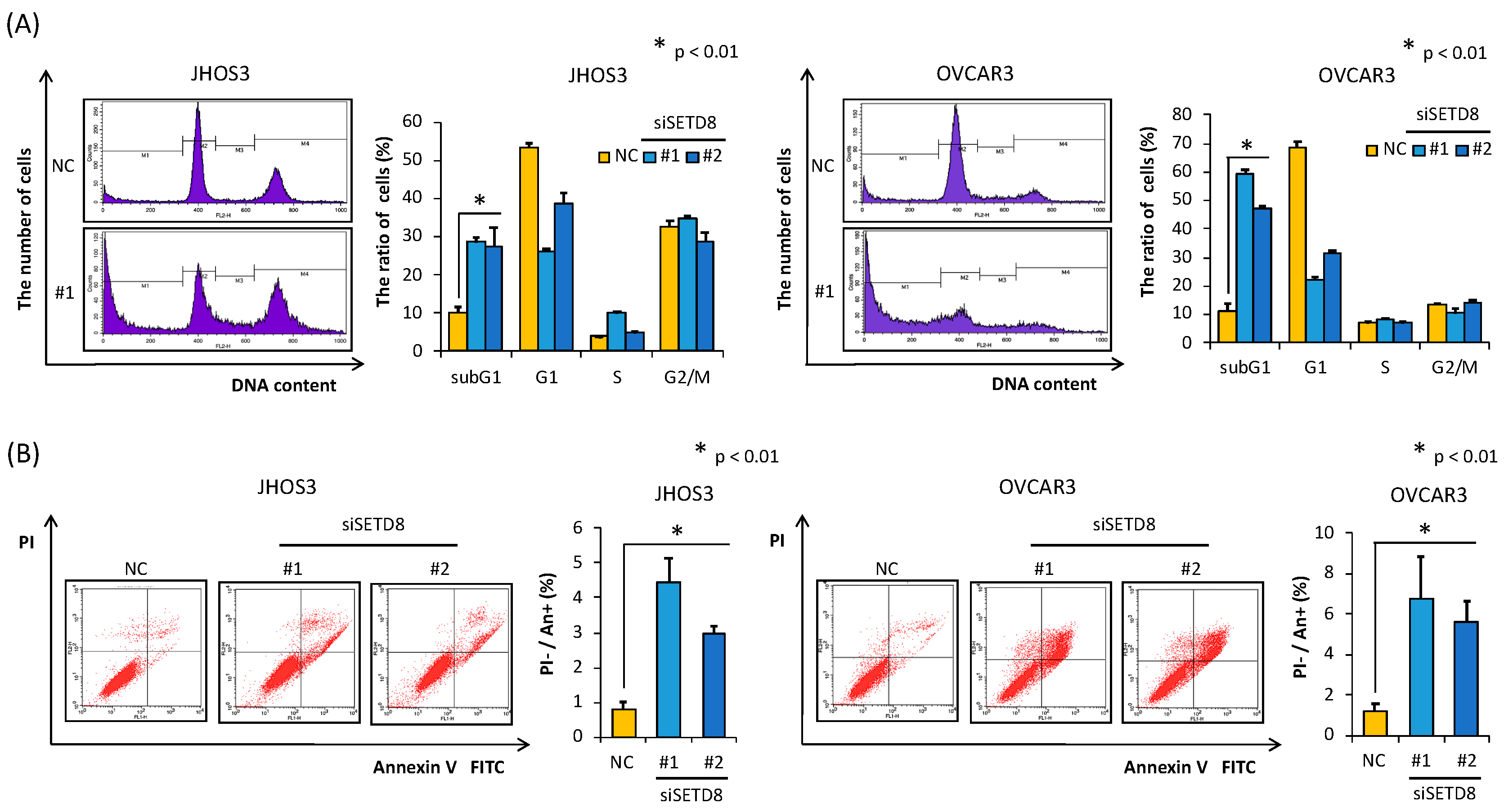
3.2. SETD8 Is Involved in the Growth of HGSOC Cells through H4K20 Monomethylation
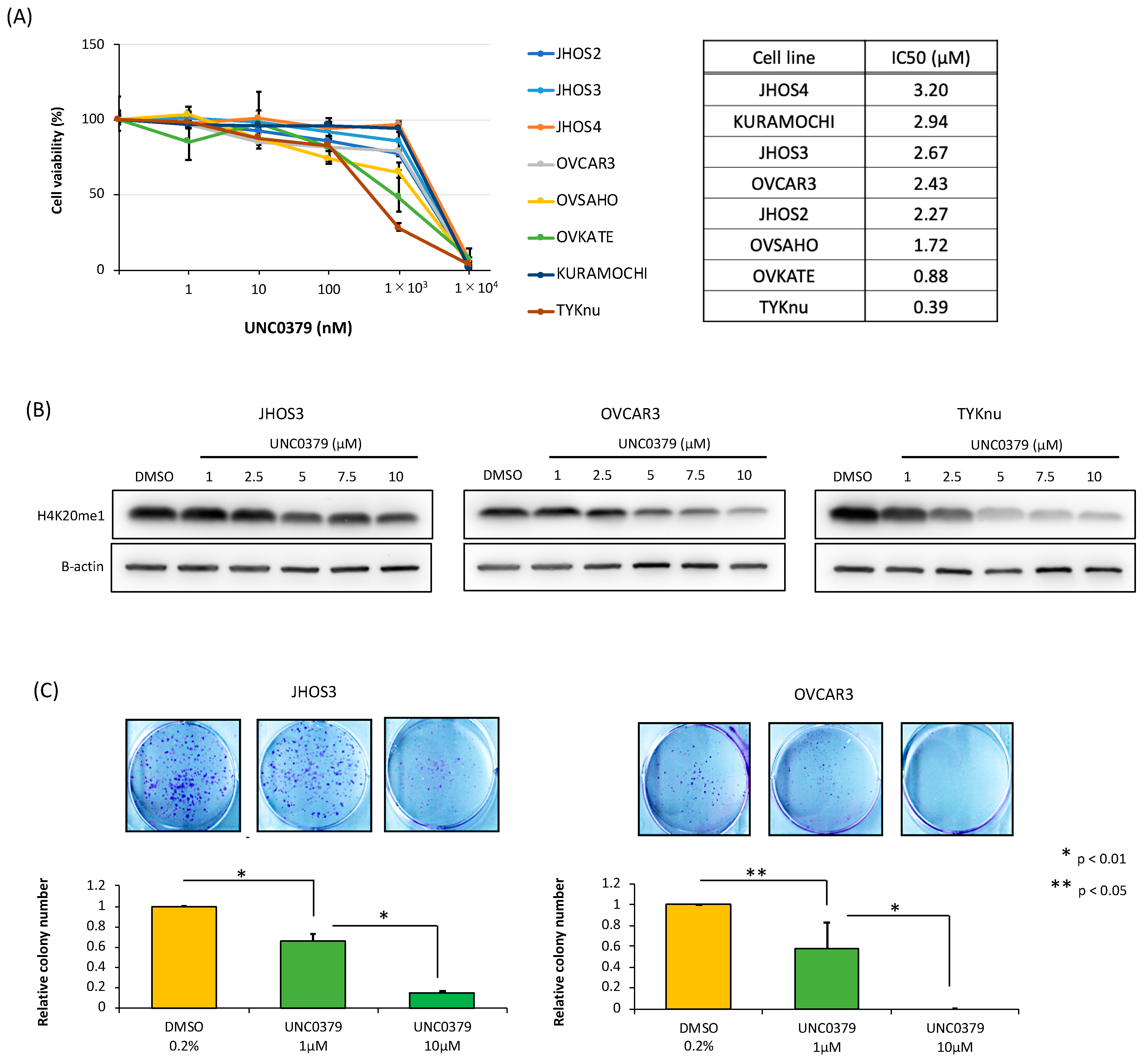
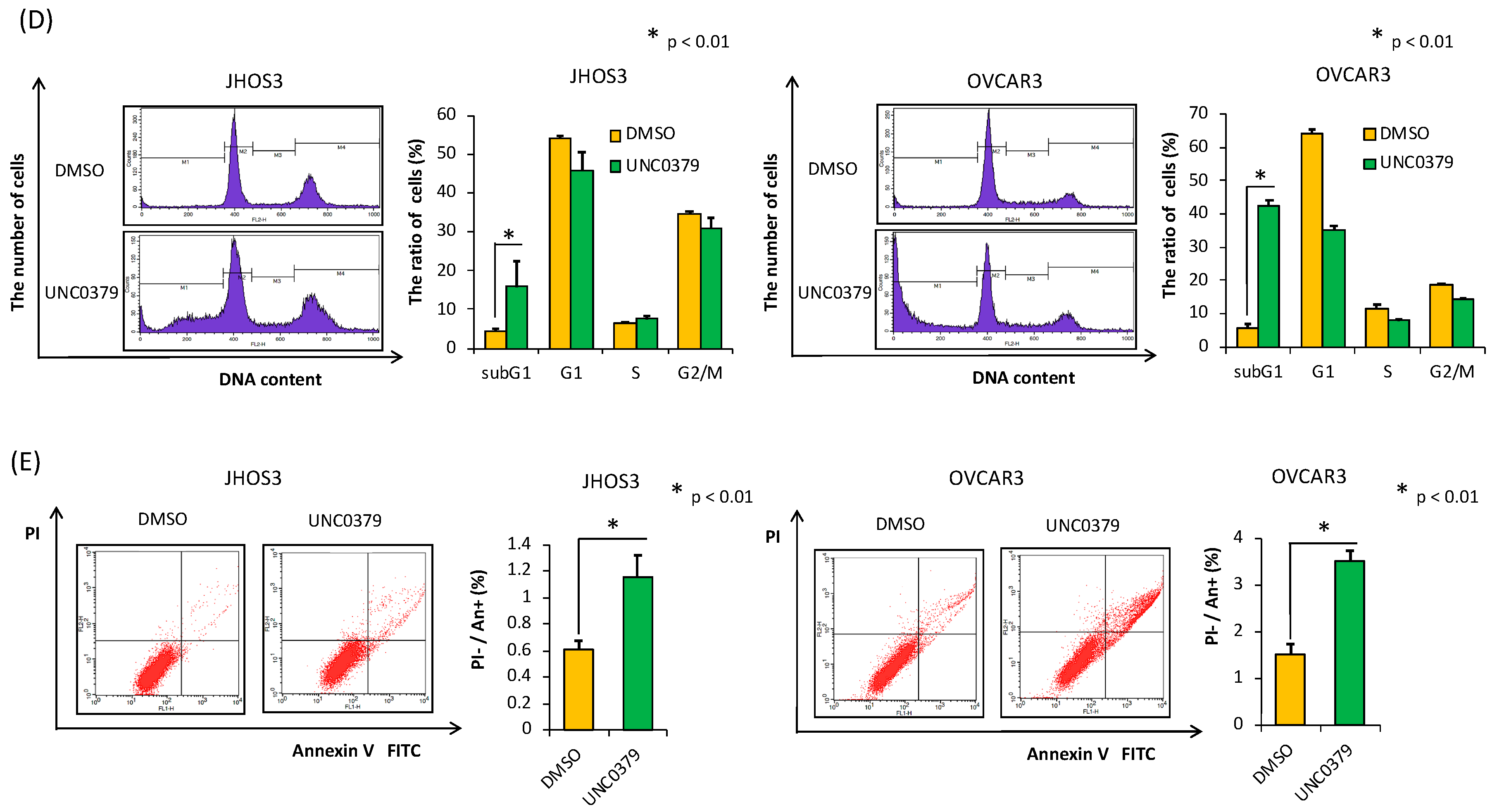
3.3. A SETD8-Selective Inhibitor Suppresses Cell Proliferation and Induces Apoptosis in HGSOC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data availability
References
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef]
- Kohn, E.C.; Ivy, S.P. Whence high-grade serous ovarian cancer. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-grade serous ovarian cancer: Basic sciences, clinical and therapeutic standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margueron, R.; Trojer, P.; Reinberg, D. The key to development: Interpreting the histone code? Curr. Opin. Genet. Dev. 2005, 15, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Varier, R.A.; Timmers, H.T.M. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef]
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nature 2015, 15, 110–124. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37. [Google Scholar] [CrossRef]
- Sone, K.; Piao, L.; Nakakido, M.; Ueda, K.; Jenuwein, T.; Nakamura, Y.; Hamamoto, R. Critical role of lysine 134 methylation on histone H2AX for γ-H2AX production and DNA repair. Nat. Commun. 2014, 5, 5691. [Google Scholar] [CrossRef] [Green Version]
- Kukita, A.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Wada, M.; Honjoh, H.; Kawata, Y.; Wada, M.; et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019, 513, 340–346. [Google Scholar] [CrossRef]
- Couture, J.F.; Collazo, E.; Brunzelle, J.S.; Trievel, R.C. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005, 19, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Feng, Q.; Ketel, C.S.; Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Simon, J.A.; Zhang, Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002, 12, 1086–1099. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takawa, M.; Cho, H.S.; Hayami, S.; Toyokawa, G.; Kogure, M.; Yamane, Y.; Iwai, Y.; Maejima, K.; Ueda, K.; Masuda, A.; et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012, 72, 3217–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhami, G.K.; Liu, H.; Galka, M.; Voss, C.; Wei, R.; Muranko, K.; Kaneko, T.; Cregan, S.P.; Li, L.; Li, S.S. Dynamic methylation of Numb by Set8 regulates its binding to p53 and apoptosis. Mol. Cell. 2013, 50, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.L.; Guo, Z.J.; Wang, X.L.; Liu, X.L.; Shi, G.F. SET8 expression is associated with overall survival in gastric cancer. Genet. Mol. Res. 2015, 14, 15609–15615. [Google Scholar] [CrossRef]
- Wang, C.; Wu, J.; Zhao, Y.; Guo, Z. MiR-502 mediated histone methyltransferase SET8 expression is associated with outcome of esophageal squamous cell carcinoma. Sci. Rep. 2016, 6, 32921. [Google Scholar] [CrossRef]
- Hou, L.; Li, Q.; Yu, Y.; Li, M.; Zhang, D. SET8 induces epithelial-mesenchymal transition and enhances prostate cancer cell metastasis by cooperating with ZEB1. Mol. Med. Rep. 2016, 13, 1681–1688. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Du, F.; Han, X.; Li, X.; Niu, Y.; Ren, S.; Sun, Y. Histone H4 Lys 20 methyltransferase SET8 promotes androgen receptor-mediated transcription activation in prostate cancer. Biochem Biophys. Res. Commun. 2014, 450, 692–696. [Google Scholar] [CrossRef]
- Veschi, V.; Liu, Z.; Voss, T.C.; Ozbun, L.; Gryder, B.; Yan, C.; Hu, Y.; Ma, A.; Jin, J.; Mazur, S.J.; et al. Epigenetic siRNA and chemical screens identify SETD8 inhibition as a therapeutic strategy for p53 activation in high-risk neuroblastoma. Cancer Cell 2017, 31, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Yu, W.; Li, F.; Bleich, R.M.; Herold, J.M.; Butler, K.V.; Norris, J.L.; Korboukh, V.; Tripathy, A.; Janzen, W.P.; et al. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 2014, 57, 6822–6833. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Xu, J.; Bai, Y.; Cui, L.; Zhang, H.; Zhang, S.L.; Zhang, D.X. Expression of SET8 protein in renal clear cell carcinoma and its clinical significance. Tumor 2017, 37, 1063–1068. [Google Scholar]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noë, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated genomic, epigenomic, and expression analyses of ovarian cancer cell lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef] [Green Version]
- Veo, B.; Danis, E.; Pierce, A.; Sola, I.; Wang, D.; Foreman, N.K.; Jin, J.; Ma, A.; Serkova, N.; Venkataraman, S.; et al. Combined functional genomic and chemical screens identify SETD8 as a therapeutic target in MYC-driven medulloblastoma. JCI Insight 2019, 4, e122933. [Google Scholar] [CrossRef]
- Liao, T.; Wang, Y.J.; Hu, J.Q.; Wang, Y.; Han, L.T.; Ma, B.; Shi, R.L.; Qu, N.; Wei, W.J.; Guan, Q.; et al. Histone methyltransferase KMT5A gene modulates oncogenesis and lipid metabolism of papillary thyroid cancer in vitro. Oncol. Rep. 2018, 39, 2185–2192. [Google Scholar] [CrossRef] [Green Version]
- Gursoy-Yuzugullu, O.; Carman, C.; Serafim, R.B.; Myronakis, M.; Valente, V.; Price, B.D. Epigenetic therapy with inhibitors of histone methylation suppresses DNA damage signaling and increases glioma cell radiosensitivity. Oncotarget 2017, 8, 24518. [Google Scholar] [CrossRef] [Green Version]
- Mosallayi, M.; Simonian, M.; Khosravi, S.; Salehi, A.R.; Khodadoostan, M.; Sebghatollahi, V.; Baradaran, A.; Salehi, R. Polymorphism (rs16917496) at the miR-502 binding site of the lysine methyltransferase 5A (SET8) and its correlation with colorectal cancer in Iranians. Adv. Biomed. Res. 2017, 6, 77. [Google Scholar]
- Hassan, N.; Rutsch, N.; Győrffy, B.; Espinoza-Sánchez, N.A.; Götte, M. SETD3 acts as a prognostic marker in breast cancer patients and modulates the viability and invasion of breast cancer cells. Sci. Rep. 2020, 10, 2262. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wada, M.; Kukita, A.; Sone, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Takahashi, Y.; Inoue, F.; Kojima, M.; Honjoh, H.; et al. Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer. Biomolecules 2020, 10, 1686. https://doi.org/10.3390/biom10121686
Wada M, Kukita A, Sone K, Hamamoto R, Kaneko S, Komatsu M, Takahashi Y, Inoue F, Kojima M, Honjoh H, et al. Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer. Biomolecules. 2020; 10(12):1686. https://doi.org/10.3390/biom10121686
Chicago/Turabian StyleWada, Miku, Asako Kukita, Kenbun Sone, Ryuji Hamamoto, Syuzo Kaneko, Masaaki Komatsu, Yu Takahashi, Futaba Inoue, Machiko Kojima, Harunori Honjoh, and et al. 2020. "Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer" Biomolecules 10, no. 12: 1686. https://doi.org/10.3390/biom10121686