Autophagy and Intracellular Membrane Trafficking Subversion by Pathogenic Yersinia Species

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy
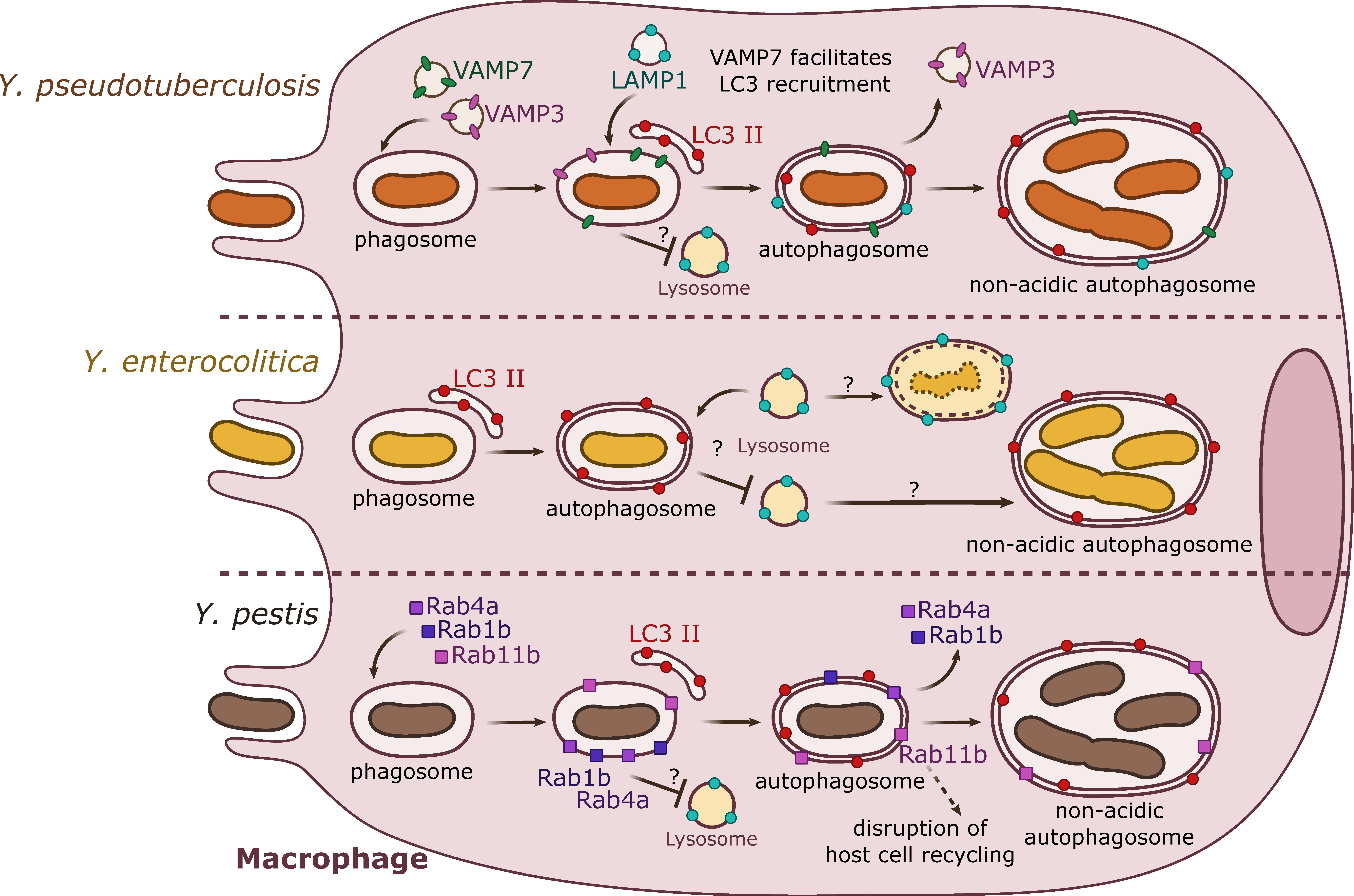
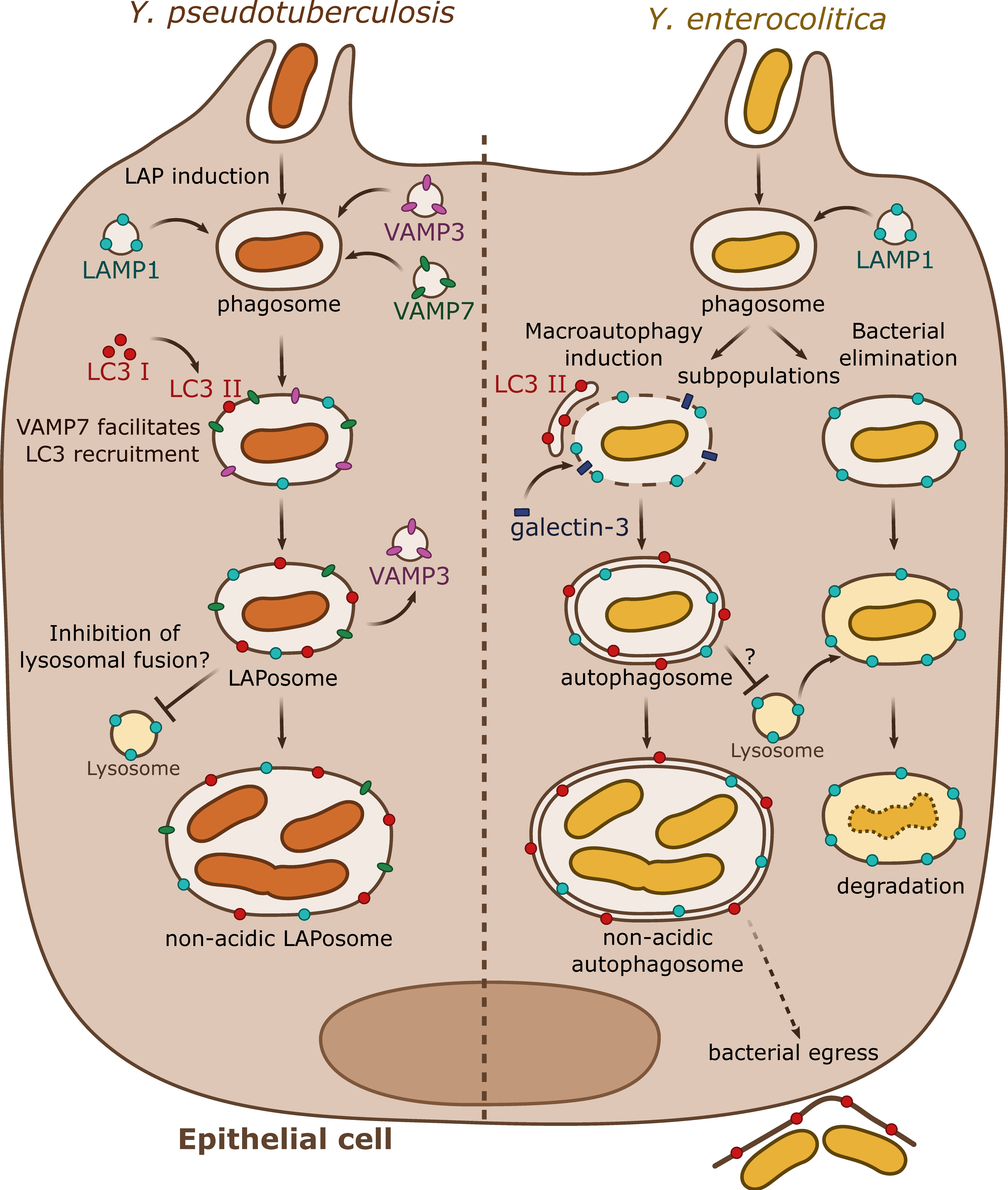
3. Yersinia pseudotuberculosis
4. Yersinia enterocolitica
5. Yersinia pestis
6. Yersinia ruckeri
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Naktin, J.; Beavis, K.G. Yersinia enterocolitica and Yersinia pseudotuberculosis. Clin. Lab. Med. 1999, 19, 523–536. [Google Scholar] [CrossRef]
- Demeure, C.E.; Dussurget, O.; Fiol, G.M.; Le Guern, A.-S.; Savin, C.; Pizarro-Cerdá, J. Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Genes Immun. 2019, 20, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, J.C.; Miller, V.L. Yersinia enterocolitica invasin: A primary role in the initiation of infection. Proc. Natl. Acad. Sci. USA 1993, 90, 6473–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, M.A.; Hirst, B.H.; Jepson, M.A. M-Cell Surface Integrin Expression and Invasin-Mediated Targeting of Yersinia pseudotuberculosis to Mouse Peyer’s Patch M Cells. Infect. Immun. 1998, 66, 1237–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahlgren, A.; Avican, K.; Westermark, L.; Nordfelth, R.; Fällman, M. Colonization of Cecum Is Important for Development of Persistent Infection by Yersinia pseudotuberculosis. Infect. Immun. 2014, 82, 3471–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottone, E.J. Yersinia enterocolitica: The charisma continues. Clin. Microbiol. Rev. 1997, 10, 257–276. [Google Scholar] [CrossRef]
- Simonet, M.; Riot, B.; Fortineau, N.; Berche, P. Invasin production by Yersinia pestis is abolished by insertion of an IS200-like element within the inv gene. Infect. Immun. 1996, 64, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Sebbane, F.; Gardner, D.; Long, D.; Gowen, B.B.; Hinnebusch, B.J. Kinetics of Disease Progression and Host Response in a Rat Model of Bubonic Plague. Am. J. Pathol. 2005, 166, 1427–1439. [Google Scholar] [CrossRef] [Green Version]
- Cornelis, G.R.; Wolf-Watz, H. The Yersinia Yop virulon: A bacterial system for subverting eukaryotic cells. Mol. Microbiol. 1997, 23, 861–867. [Google Scholar] [CrossRef]
- Cornelis, G.R. The Yersinia Ysc-Yop “type III” weaponry. Nat. Rev. Mol. Cell Biol. 2002, 3, 742–752. [Google Scholar] [CrossRef]
- Isberg, R.R.; Falkow, S. A single genetic locus encoded by Yersinia pseudotuberculosis permits invasion of cultured animal cells by Escherichia coli K-12. Nature 1985, 317, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Isberg, R.R.; Voorhis, D.L.; Falkow, S. Identification of invasin: A protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 1987, 50, 769–778. [Google Scholar] [CrossRef]
- Isberg, R.R.; Leong, J.M. Multiple beta β1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 1990, 60, 861–871. [Google Scholar] [CrossRef]
- Guinet, F.; Avé, P.; Filali, S.; Huon, C.; Savin, C.; Huerre, M.; Fiette, L.; Carniel, E. Dissociation of Tissue Destruction and Bacterial Expansion during Bubonic Plague. PLoS Pathogens 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Pujol, C.; Bliska, J.B. The Ability To Replicate in Macrophages Is Conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 2003, 71, 5892–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabenstein, J.P.; Marceau, M.; Pujol, C.; Simonet, M.; Bliska, J.B. The Response Regulator PhoP of Yersinia pseudotuberculosis Is Important for Replication in Macrophages and for Virulence. Infect. Immun. 2004, 72, 4973–4984. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.; Rusten, T.E. Origin and evolution of self-consumption: Autophagy. Adv. Exp. Med. Biol. 2007, 607, 111–118. [Google Scholar] [CrossRef]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Aoki, A.; Kusabiraki, T.; Shima, T.; Yoshino, O.; Cheng, S.-B.; Sharma, S.; Saito, S. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J. Obstet. Gynaecol. Res. 2017, 43, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, S.; Philips, J.A. LC3-associated phagocytosis: Host defense and microbial response. Curr. Opin. Immunol. 2019, 60, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Grimmel, M.; Backhaus, C.; Proikas-Cezanne, T. WIPI-Mediated Autophagy and Longevity. Cells 2015, 4, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y.; Mizushima, N. Two ubiquitin-like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 2004, 15, 231–236. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Hayashi, K.; Taura, M.; Iwasaki, A. The interaction between IKKα and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci. Signal. 2018, 11, 11–21. [Google Scholar] [CrossRef]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.O.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [Green Version]
- Herb, M.; Gluschko, A.; Schramm, M. LC3-associated phagocytosis—The highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 2020, 101, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shpilka, T.; Shvets, E.; Abada, A.; Shimron, F.; Elazar, Z. LC3 and GATE-16 N Termini mediate membrane fusion processes required for autophagosome biogenesis. Dev. Cell 2011, 20, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.I.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef]
- Moreau, K.; Lacas-Gervais, S.; Fujita, N.; Sebbane, F.; Yoshimori, T.; Simonet, M.; Lafont, F. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell. Microbiol. 2010, 12, 1108–1123. [Google Scholar] [CrossRef]
- Tsukano, H.; Kura, F.; Inoue, S.; Sato, S.; Izumiya, H.; Yasuda, T.; Watanabe, H. Yersinia pseudotuberculosis blocks the phagosomal acidification of B10.A mouse macrophages through the inhibition of vacuolar H+-ATPase activity. Microb. Pathog. 1999, 27, 253–263. [Google Scholar] [CrossRef]
- Ligeon, L.-A.; Moreau, K.; Barois, N.; Bongiovanni, A.; Lacorre, D.-A.; Werkmeister, E.; Proux-Gillardeaux, V.; Galli, T.; Lafont, F. Role of VAMP3 and VAMP7 in the commitment of Yersinia pseudotuberculosis to LC3-associated pathways involving single- or double-membrane vacuoles. Autophagy 2014, 10, 1588–1602. [Google Scholar] [CrossRef] [Green Version]
- Deuretzbacher, A.; Czymmeck, N.; Reimer, R.; Trülzsch, K.; Gaus, K.; Hohenberg, H.; Heesemann, J.; Aepfelbacher, M.; Ruckdeschel, K. β 1 Integrin-Dependent Engulfment of Yersinia enterocolitica by Macrophages Is Coupled to the Activation of Autophagy and Suppressed by Type III Protein Secretion. J. Immunol. 2009, 183, 5847–5860. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.G.; Pulsifer, A.R.; Price, C.T.; Abu Kwaik, Y.; Lawrenz, M.B. Yersinia pestis requires host Rab1b for survival in macrophages. PLoS Pathog 2015, 11, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.J.V.; Schimmeck, H.; Gropengießer, J.; Middendorf, L.; Quitmann, M.; Schneider, C.; Holstermann, B.; Wacker, R.; Heussler, V.; Reimer, R.; et al. Activation of the macroautophagy pathway by Yersinia enterocolitica promotes intracellular multiplication and egress of Yersiniae from epithelial cells. Cell. Microbiol. 2019, 21, 1–18. [Google Scholar] [CrossRef]
- Charnetzky, W.T.; Shuford, W.W. Survival and growth of Yersinia pestis within macrophages and an effect of the loss of the 47-megadalton plasmid on growth in macrophages. Infect. Immun. 1985, 47, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straley, S.C.; Harmon, P.A. Yersinia pestis grows within phagolysosomes in mouse peritoneal macrophages. Infect. Immun. 1984, 45, 655–659. [Google Scholar] [CrossRef] [Green Version]
- Pujol, C.; Klein, K.A.; Romanov, G.A.; Palmer, L.E.; Cirota, C.; Zhao, Z.; Bliska, J.B. Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 2009, 77, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Birmingham, C.L.; Shahnazari, S.; Shiu, J.; Zheng, Y.T.; Smith, A.C.; Campellone, K.G.; Heo, W.D.; Gruenheid, S.; Meyer, T.; et al. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy 2011, 7, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.G.; Pulsifer, A.R.; Chung, D.; Rouchka, E.C.; Ceresa, B.K.; Lawrenz, M.B. Yersinia pestis Targets the Host Endosome Recycling Pathway during the Biogenesis of the Yersinia-Containing Vacuole To Avoid Killing by Macrophages. mBio 2018, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.; Menanteau-Ledouble, S.; Saleh, M.; El-Matbouli, M. Yersinia ruckeri, the causative agent of enteric redmouth disease in fish. Veter. Res. 2015, 46, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kawula, T.H.; Lelivelt, M.J.; Orndorff, P.E. Using a new inbred fish model and cultured fish tissue cells to study Aeromonas hydrophila and Yersinia ruckeri pathogenesis. Microb. Pathog. 1996, 20, 119–125. [Google Scholar] [CrossRef]
- Tobback, E.; Decostere, A.; Hermans, K.; Van den Broeck, W.; Haesebrouck, F.; Chiers, K. In vitro markers for virulence in Yersinia ruckeri. J. Fish Dis. 2010, 33, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Menanteau-Ledouble, S.; Lawrence, M.L.; El-Matbouli, M. Invasion and replication of Yersinia ruckeri in fish cell cultures. BMC Veter. Res. 2018, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckaert, J.; Bossier, P.; D’Herde, K.; Diez-Fraile, A.; Sorgeloos, P.; Haesebrouck, F.; Pasmans, F. Persistence of Yersinia ruckeri in trout macrophages. Fish. Shellfish. Immunol. 2010, 29, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Oyston, P.C.F.; Dorrell, N.; Williams, K.; Li, S.-R.; Green, M.; Titball, R.W.; Wren, B.W. The Response Regulator PhoP Is Important for Survival under Conditions of Macrophage-Induced Stress and Virulence in Yersinia pestis. Infect. Immun. 2000, 68, 3419–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titball, R.W.; Hill, J.; Lawton, D.G.; Brown, K.A. Yersinia pestis and plague. Biochem. Soc. Trans. 2003, 31, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, R.J.; Lane, M.C.; Wagner, N.J.; Weening, E.H.; Miller, V.L. Dissemination of a Highly Virulent Pathogen: Tracking The Early Events That Define Infection. PLoS Pathog. 2015, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, J.G.; Bosio, C.F.; Hinnebusch, B.J. Dermal Neutrophil, Macrophage and Dendritic Cell Responses to Yersinia pestis Transmitted by Fleas. PLoS Pathog. 2015, 11, e1004734. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.J.; Miller, V.L. A Deadly Path: Bacterial Spread During Bubonic Plague. Trends. Microbiol. 2016, 24, 239–241. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.A.; Maltez, V.I.; Hall, J.D.; Vitko, N.P.; Miller, V.L. A Phenotype at Last: Essential Role for the Yersinia enterocolitica Ysa Type III Secretion System in a Drosophila melanogaster S2 Cell Model. Infect. Immun. 2013, 81, 2478–2487. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wang, K.-Y.; Wang, J.; Chen, D.-F.; Huang, X.-L.; Ouyang, P.; Geng, Y.; He, Y.; Zhou, Y.; Min, J. Genome Sequence of the Fish Pathogen Yersinia ruckeri SC09 Provides Insights into Niche Adaptation and Pathogenic Mechanism. Int. J. Mol. Sci. 2016, 17, 557. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemarignier, M.; Pizarro-Cerdá, J. Autophagy and Intracellular Membrane Trafficking Subversion by Pathogenic Yersinia Species. Biomolecules 2020, 10, 1637. https://doi.org/10.3390/biom10121637
Lemarignier M, Pizarro-Cerdá J. Autophagy and Intracellular Membrane Trafficking Subversion by Pathogenic Yersinia Species. Biomolecules. 2020; 10(12):1637. https://doi.org/10.3390/biom10121637
Chicago/Turabian StyleLemarignier, Marion, and Javier Pizarro-Cerdá. 2020. "Autophagy and Intracellular Membrane Trafficking Subversion by Pathogenic Yersinia Species" Biomolecules 10, no. 12: 1637. https://doi.org/10.3390/biom10121637