TORC2-Dependent Ypk1-Mediated Phosphorylation of Lam2/Ltc4 Disrupts Its Association with the β-Propeller Protein Laf1 at Endoplasmic Reticulum-Plasma Membrane Contact Sites in the Yeast Saccharomyces cerevisiae


Abstract
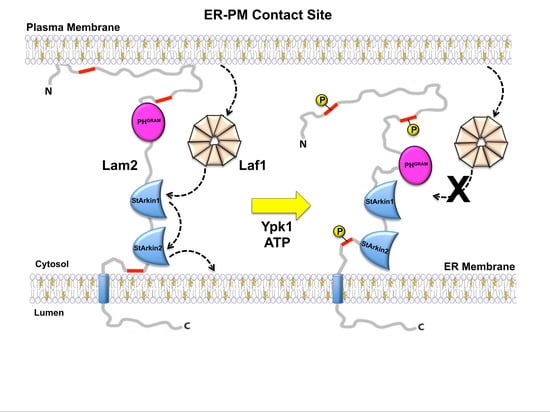
:
1. Introduction
2. Materials and Methods
2.1. Construction of Yeast Strains and Growth Conditions
2.2. Plasmids and Recombinant DNA Methods
2.3. Production and Purification of GST-Fusion Proteins
2.4. Production and Purification of Ypk1 from S. cerevisiae
2.5. Preparation of Ypk1-Phosphorylated Lam2 Fragment
2.6. In Vitro Binding Assay
2.7. Immunoblotting
2.8. Fluorescence Microscopy
3. Results
3.1. Localization of β-Propeller Proteins Laf1 and Dgr2 at ER-PM CSs Requires Lam2 and Lam4
3.2. Laf1 Is Required for Efficient Retrograde Removal of PM Ergosterol
3.3. Laf1 Physically Associates with Lam2 and the PH Domain of Lam2 Is Not Required for Its Interaction with Laf1
3.4. Ypk1-Mediated Phosphorylation of Lam2 Disrupts Its Association with GST-Laf1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zinser, E.; Paltauf, F.; Daum, G. Sterol composition of yeast organelle membranes and subcellular distribution of enzymes involved in sterol metabolism. J. Bacteriol. 1993, 175, 2853–2858. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasopoulos, A.; André, B.; Sophianopoulou, V.; Gournas, C. Fungal plasma membrane domains. FEMS Microbiol. Rev. 2019, 43, 642–673. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J. Sterols and membrane dynamics. J. Chem. Biol. 2008, 1, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, L.M.; Konopka, J.B. Fungal membrane organization: The eisosome concept. Annu. Rev. Microbiol. 2014, 68, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.L. The Multifunctional Fungal Ergosterol. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Layer, J.V.; Barnes, B.M.; Yamasaki, Y.; Barbuch, R.; Li, L.; Taramino, S.; Balliano, G.; Bard, M. Characterization of a mutation that results in independence of oxidosqualene cyclase (Erg7) activity from the downstream 3-ketoreductase (Erg27) in the yeast ergosterol biosynthetic pathway. Biochim. Biophys. Acta 2013, 1831, 361–369. [Google Scholar] [CrossRef]
- Baumann, N.A.; Sullivan, D.P.; Ohvo-Rekilä, H.; Simonot, C.; Pottekat, A.; Klaassen, Z.; Beh, C.T.; Menon, A.K. Transport of newly synthesized sterol to the sterol-enriched plasma membrane occurs via nonvesicular equilibration. Biochemistry 2005, 44, 5816–5826. [Google Scholar] [CrossRef]
- Sullivan, D.P.; Ohvo-Rekilä, H.; Baumann, N.A.; Beh, C.T.; Menon, A.K. Sterol trafficking between the endoplasmic reticulum and plasma membrane in yeast. Biochem. Soc. Trans. 2006, 34, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Mesmin, B.; Antonny, B.; Drin, G. Insights into the mechanisms of sterol transport between organelles. Cell Mol. Life Sci 2013, 70, 3405–3421. [Google Scholar] [CrossRef]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef] [PubMed]
- Gatta, A.T.; Wong, L.H.; Sere, Y.Y.; Calderón-Noreña, D.M.; Cockcroft, S.; Menon, A.K.; Levine, T.P. A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.; Sarsam, R.D.; Toulmay, A.; Yamada, J.; Prinz, W.A.; Nunnari, J. Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J. Cell Biol. 2015, 209, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; Levine, T.P. Lipid transfer proteins do their thing anchored at membrane contact sites… but what is their thing? Biochem. Soc. Trans. 2016, 44, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Tsujishita, Y.; Hurley, J.H. Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 2000, 7, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, P.; Najmanivich, R.; Lehoux, J.G. Mammalian StAR-related lipid transfer (START) domains with specificity for cholesterol: Structural conservation and mechanism of reversible binding. Subcell. Biochem. 2010, 51, 425–437. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Iaea, D.B.; Pipalia, N.H. Role of STARD4 and NPC1 in intracellular sterol transport. Biochem. Cell Biol. 2016, 94, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Manik, M.K.; Im, Y.J. Structural basis of sterol recognition and nonvesicular transport by lipid transfer proteins anchored at membrane contact sites. Proc. Natl. Acad. Sci. USA 2018, 115, E856–E865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horenkamp, F.A.; Valverde, D.P.; Nunnari, J.; Reinisch, K.M. Molecular basis for sterol transport by StART-like lipid transfer domains. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, J.A.; Kiburu, I.; Pandey, K.; Timme, M.; Ramlall, T.; Levkau, B.; Wu, J.; Eliezer, D.; Boudker, O.; Menon, A.K. Structural basis of sterol binding and transport by a yeast StARkin domain. J. Biol. Chem. 2018, 293, 5522–5531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, H.; Gaigg, B.; Hrastnik, C.; Achleitner, G.; Kohlwein, S.D.; Zellnig, G.; Perktold, A.; Daum, G. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur. J. Biochem. 2001, 268, 2351–2361. [Google Scholar] [CrossRef]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef]
- Gatta, A.T.; Sauerwein, A.C.; Zhuravleva, A.; Levine, T.P.; Matthews, S. Structural insights into a StART-like domain in Lam4 and its interaction with sterol ligands. Biochem. Biophys. Res. Commun. 2018, 495, 2270–2274. [Google Scholar] [CrossRef] [Green Version]
- Khelashvili, G.; Chauhan, N.; Pandey, K.; Eliezer, D.; Menon, A.K. Exchange of water for sterol underlies sterol egress from a StARkin domain. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zinser, E.; Sperka-Gottlieb, C.D.; Fasch, E.V.; Kohlwein, S.D.; Paltauf, F.; Daum, G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 1991, 173, 2026–2034. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef]
- Pemberton, J.G.; Balla, T. Polyphosphoinositide-Binding Domains: Insights from Peripheral Membrane and Lipid-Transfer Proteins. Adv. Exp. Med. Biol. 2019, 1111, 77–137. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Mendrola, J.M.; Audhya, A.; Singh, S.; Keleti, D.; DeWald, D.B.; Murray, D.; Emr, S.D.; Lemmon, M.A. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 2004, 13, 677–688. [Google Scholar] [CrossRef]
- Bartlett, K.; Kim, K. Insight into Tor2, a budding yeast microdomain protein. Eur. J. Cell Biol. 2014, 93, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Leskoske, K.L.; Martinez Marshall, M.N.; Locke, M.N.; Thorner, J. The TORC2-Dependent Signaling Network in the Yeast Saccharomyces cerevisiae. Biomolecules 2017, 7, 66. [Google Scholar] [CrossRef]
- Riggi, M.; Kusmider, B.; Loewith, R. The flipside of the TOR coin—TORC2 and plasma membrane homeostasis at a glance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Muir, A.; Ramachandran, S.; Roelants, F.M.; Timmons, G.; Thorner, J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Roelants, F.M.; Chauhan, N.; Muir, A.; Davis, J.C.; Menon, A.K.; Levine, T.P.; Thorner, J. TOR complex 2-regulated protein kinase Ypk1 controls sterol distribution by inhibiting StARkin domain-containing proteins located at plasma membrane-endoplasmic reticulum contact sites. Mol. Biol. Cell 2018, 29, 2128–2136. [Google Scholar] [CrossRef]
- Murley, A.; Yamada, J.; Niles, B.J.; Toulmay, A.; Prinz, W.A.; Powers, T.; Nunnari, J. Sterol transporters at membrane contact sites regulate TORC1 and TORC2 signaling. J. Cell Biol. 2017, 216, 2679–2689. [Google Scholar] [CrossRef] [Green Version]
- Stirnimann, C.U.; Petsalaki, E.; Russell, R.B.; Müller, C.W. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010, 35, 565–574. [Google Scholar] [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.R.; Kemmeren, P.; Zhao, X.C.; Greenblatt, J.F.; Spencer, F.; Holstege, F.C.; Weissman, J.S.; Krogan, N.J. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell Proteom. 2007, 6, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Amberg, D.C.; Burke, D.J.; Strathern, J.N. Methods in Yeast Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2005. [Google Scholar]
- Alvaro, C.G.; O’Donnell, A.F.; Prosser, D.C.; Augustine, A.A.; Goldman, A.; Brodsky, J.L.; Cyert, M.S.; Wendland, B.; Thorner, J. Specific α-arrestins negatively regulate Saccharomyces cerevisiae pheromone response by down-modulating the G-protein-coupled receptor Ste2. Mol. Cell Biol. 2014, 34, 2660–2681. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.S.; Klionsky, D.J.; Banta, L.M.; Emr, S.D. Protein sorting in Saccharomyces cerevisiae: Isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell Biol. 1988, 8, 4936–4948. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; Macgurn, J.A.; Emr, S.D. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev. Cell 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012; Volume 1–2. [Google Scholar]
- Roelants, F.M.; Breslow, D.K.; Muir, A.; Weissman, J.S.; Thorner, J. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2011, 108, 19222–19227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A. Systematic Identification of Proteins Regulated by the TOR Complex. 2-Dependent Kinase Ypk1 in Saccharomyces Cerevisiae; University of California: Berkeley, CA, USA, 2015; p. 123. [Google Scholar]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Baum, P.; Thorner, J.; Honig, L. Identification of tubulin from the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1978, 75, 4962–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelstein, A.D.; Amodaj, N.; Hoover, K.H.; Vale, R.D.; Stuurman, N. Computer Control of Microscopes Using µManager. Curr. Protoc. Mol. Biol. 2010, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, P.C.; Bharat, T.A.M.; Wozny, M.R.; Boulanger, J.; Miller, E.A.; Kukulski, W. Tricalbins Contribute to Cellular Lipid Flux and Form Curved ER-PM Contacts that Are Bridged by Rod-Shaped Structures. Dev. Cell 2019, 51, 488–502.e488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quon, E.; Sere, Y.Y.; Chauhan, N.; Johansen, J.; Sullivan, D.P.; Dittman, J.S.; Rice, W.J.; Chan, R.B.; Di Paolo, G.; Beh, C.T.; et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018, 16, e2003864. [Google Scholar] [CrossRef] [Green Version]
- Gururaj, C.; Federman, R.S.; Federman, R.; Chang, A. Orm proteins integrate multiple signals to maintain sphingolipid homeostasis. J. Biol. Chem. 2013, 288, 20453–20463. [Google Scholar] [CrossRef] [Green Version]
- Plank, M.; Perepelkina, M.; Müller, M.; Vaga, S.; Zou, X.; Bourgoint, C.; Berti, M.; Saarbach, J.; Haesendonckx, S.; Winssinger, N.; et al. Chemical Genetics of AGC-kinases Reveals Shared Targets of Ypk1, Protein Kinase A and Sch9. Mol. Cell Proteom. 2020, 19, 655–671. [Google Scholar] [CrossRef] [Green Version]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.; Baryshnikova, A.; Brown, G.W. Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell Syst. 2018, 6, 192–205.e193. [Google Scholar] [CrossRef] [Green Version]
- Locke, M.N.; Thorner, J. Rab5 GTPases are required for optimal TORC2 function. J. Cell Biol. 2019, 218, 961–976. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, F.; Zhuo, Z.; Wu, X.H.; Wu, Y.D. A method for WD40 repeat detection and secondary structure prediction. PLoS ONE 2013, 8, e65705. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hu, X.J.; Zou, X.D.; Wu, X.H.; Ye, Z.Q.; Wu, Y.D. WDSPdb: A database for WD40-repeat proteins. Nucleic Acids Res. 2015, 43, D339–D344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, H.K.; Hurley, J.B.; Hopkins, R.S.; Miake-Lye, R.; Johnson, M.S.; Doolittle, R.F.; Simon, M.I. Repetitive segmental structure of the transducin beta subunit: Homology with the CDC4 gene and identification of related mRNAs. Proc. Natl. Acad. Sci. USA 1986, 83, 2162–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.K.; Chan, N.L.; Wang, A.H. The many blades of the β-propeller proteins: Conserved but versatile. Trends Biochem. Sci. 2011, 36, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Madsen, P.; Grøftehauge, M.K.; Nissen, P.; Petersen, C.M.; Thirup, S.S. Ligands bind to Sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat. Struct. Mol. Biol. 2009, 16, 96–98. [Google Scholar] [CrossRef]
- Chauhan, N.; Sere, Y.Y.; Sokol, A.M.; Graumann, J.; Menon, A.K. A PhotoClick cholesterol-based quantitative proteomics screen for cytoplasmic sterol-binding proteins in Saccharomyces cerevisiae. Yeast 2020, 37, 15–25. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Source/Reference |
|---|---|---|
| BY4741 | MATahis3∆1 leu2∆0 met15∆0 ura3∆0 | Research Genetics, Inc. (Huntsville, AL, USA) |
| BY4742 | MAT his3∆1 leu2∆0 lys2∆0 ura3∆0 | Research Genetics, Inc. |
| YFR651-A | BY4741 Laf1-mKate::SpHIS5 | This study |
| YFR652-B | BY4742 Laf1-mKate::SpHIS5 lam2∆::HygMX | This study |
| YFR653-B | BY4742 Laf1-mKate::SpHIS5 lam4∆::KanMX | This study |
| YFR654-B | BY4742 Laf1-mKate::SpHIS5 lam2∆::HygMX lam4∆:: KanMX | This study |
| YFR660 | BY4741 Ypk1(L424A)::ura3- ypk2∆::KanMX Laf1-mKate::SpHIS5 | This study |
| YFR657-A | BY4741 Dgr2-mKate:: SpHIS5 | This study |
| YFR657-B | BY4742 Dgr2-mKate:: SpHIS5 | This study |
| YFR658-B | BY4742 Dgr2-mKate:: SpHIS5 lam2∆::HygMX | This study |
| YFR659-B | BY4742 Dgr2-mKate:: SpHIS5 lam4∆::KanMX | This study |
| YFR650-B | BY4742 Dgr2-mKate:: SpHIS5 lam2∆::HygMX lam4∆:: KanMX | This study |
| YFR512-A | BY4741 GFP-Lam2::URA3 | [37] |
| YFR613-A | BY4742 GFP-Lam2::URA3 laf1∆::KanMX dgr2∆::KanMX | This study |
| YFR513 | BY4741 lam2∆:: HygMX lam4∆::KanMX | [37] |
| YFR633-A | BY4741 GFP-Lam2::URA3 Laf1-mKate::SpHIS5 | This study |
| YFR726-A | BY4742 GFP-Lam2::URA3 Laf1-mKate::SpHIS5 | This study |
| YFR635 | BY4742 Laf1-mKate::SpHIS5 | This study |
| YFR728 | BY4742 Laf1-mKate GFP-Lam2::URA3 lam4∆::KanMX | This study |
| YFR721 | BY4742 Laf1-mKate::SpHis5 GFP-Lam2(S44A T45A T518A L519A T1237A V1238A)::URA3 lam4∆::KanMX | This study |
| YFR722 | BY4741 Laf1-mKate::SpHis5 GFP-Lam2(S44E T45E T518E L519E T1237E V1238E)::URA3 lam4∆::KanMX | This study |
| yEPS23 | BY4741 GFP-Lam2::URA3 Laf1-mKate::SpHIS5 Pil1-BFP::KanMX | This study |
| YFR713 | BY4741 Laf1-mNG::LEU2 | This study |
| YFR714 | BY4741 Laf1-mNG::LEU2 lam2∆:: HygMX | This study |
| YFR715 | BY4742 Laf1-mNG::LEU2 lam4∆:: KanMX | This study |
| YFR720 | BY4742 Laf1-mNG::LEU2 lam2∆::HygMX lam4∆::KanMX | This study |
| ymr102c∆ | BY4742 laf1∆::KanMX | Research Genetics, Inc. |
| YFR734-A | laf1∆::KanMX lam2∆::HygMX lam4∆::KanMX | This study |
| YFR603 | BY4741 GFP-Lam2::URA3 laf1∆::KanMX | This study |
| YFR607 | BY4741 GFP-Lam2::URA3 dgr2∆::KanMX | This study |
| CGA84 | MATaleu2∆1::GEV::NATMX pep4∆::HIS3 prb1∆1.6R ura3-52 trp1-1 lys2-801a leu2∆1 his3∆200 can1 GAL | [46] |
| SEY6210 | MAT leu2-3,112 ura3-52 his3∆200 trp1∆901 suc2∆9 lys2-801a GAL | [47] |
| YFR712 | SEY6210 Lam2-mNG::LEU2 | This study |
| ANDY198 | SEY6210 ist2∆::hisMX6 scs2∆::TRP1 scs22∆::hisMX6 tcb1∆::kanMX6 tcb2∆::kanMX6 tcb3∆::hisMX6 | [48] |
| YFR704 | ANDY198 Lam2-mNG::LEU2 | This study |
| YFR729 | SEY6210 Laf1-mNG::LEU2 | This study |
| YFR705 | ANDY198 Laf1-mNG::LEU2 | This study |
| Plasmid | Description | Source/Reference |
|---|---|---|
| pGEX4T-1 | GST tag, bacterial expression vector | GE Healthcare, Inc. |
| pFR398 | pGEX4T-1 Laf1(684–834) | This study |
| pFR402 | pGEX4T-1 Laf1(684–834) S709S S710A | This study |
| pFR399 | pGEX4T-1 Dgr2(1–128) | This study |
| pFR403 | pGEX4T-1 Dgr2(1–128) S47A S48A | This study |
| pFR203 | pGEX4T-1 Orm1(1–85) | [50] |
| pMT2 | pGEX4T-1 Laf1 | This study |
| pFR396 | pGEX4T-1 Laf1(S708A S709A) | This study |
| pFR404 | pGEX4T-1 Dgr2 | This study |
| pKM263 | GST tag, bacterial expression vector | |
| pFR391 | pKM263-Lam2(482–1259)-His6 | This study |
| pFR392 | pKM263-Lam2(482–1259) T518A T1237A-His6 | This study |
| pFR393 | pKM263-Lam2(482–1259)∆PH(644–760)-His6 | This study |
| pFR407 | pKM263-Lam4(371–1177)-His6 | This study |
| pFR408 | pKM263-Lam2(633–1259)-His6 | This study |
| pFR409 | pKM263-Lam2(849–1259)-His6 | This study |
| pGEX6P-1 | GST tag, bacterial expression vector | GE Healthcare, Inc. |
| pFR377 | pGEX6P-1 Lam2(482–1259) | This study |
| pFR411 | pGEX6P-1 Lam2(482–1259) T518A L519A T1237A V1238A-His6 | This study |
| pFR413 | pGEX6P-1 Lam2(482–1259) T518E L519E T1237E V1238E-His6 | This study |
| BG1805 | C-terminal His6-HA-3C-ZZ tag, yeast expression vector | GE Healthcare, Inc. |
| pJT4317 | BG1805 Ypk1-His6-HA-3C-ZZ | GE Healthcare, Inc. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Topolska, M.; Roelants, F.M.; Si, E.P.; Thorner, J. TORC2-Dependent Ypk1-Mediated Phosphorylation of Lam2/Ltc4 Disrupts Its Association with the β-Propeller Protein Laf1 at Endoplasmic Reticulum-Plasma Membrane Contact Sites in the Yeast Saccharomyces cerevisiae. Biomolecules 2020, 10, 1598. https://doi.org/10.3390/biom10121598
Topolska M, Roelants FM, Si EP, Thorner J. TORC2-Dependent Ypk1-Mediated Phosphorylation of Lam2/Ltc4 Disrupts Its Association with the β-Propeller Protein Laf1 at Endoplasmic Reticulum-Plasma Membrane Contact Sites in the Yeast Saccharomyces cerevisiae. Biomolecules. 2020; 10(12):1598. https://doi.org/10.3390/biom10121598
Chicago/Turabian StyleTopolska, Magdalena, Françoise M. Roelants, Edward P. Si, and Jeremy Thorner. 2020. "TORC2-Dependent Ypk1-Mediated Phosphorylation of Lam2/Ltc4 Disrupts Its Association with the β-Propeller Protein Laf1 at Endoplasmic Reticulum-Plasma Membrane Contact Sites in the Yeast Saccharomyces cerevisiae" Biomolecules 10, no. 12: 1598. https://doi.org/10.3390/biom10121598