PP2A-B55 Holoenzyme Regulation and Cancer


Abstract
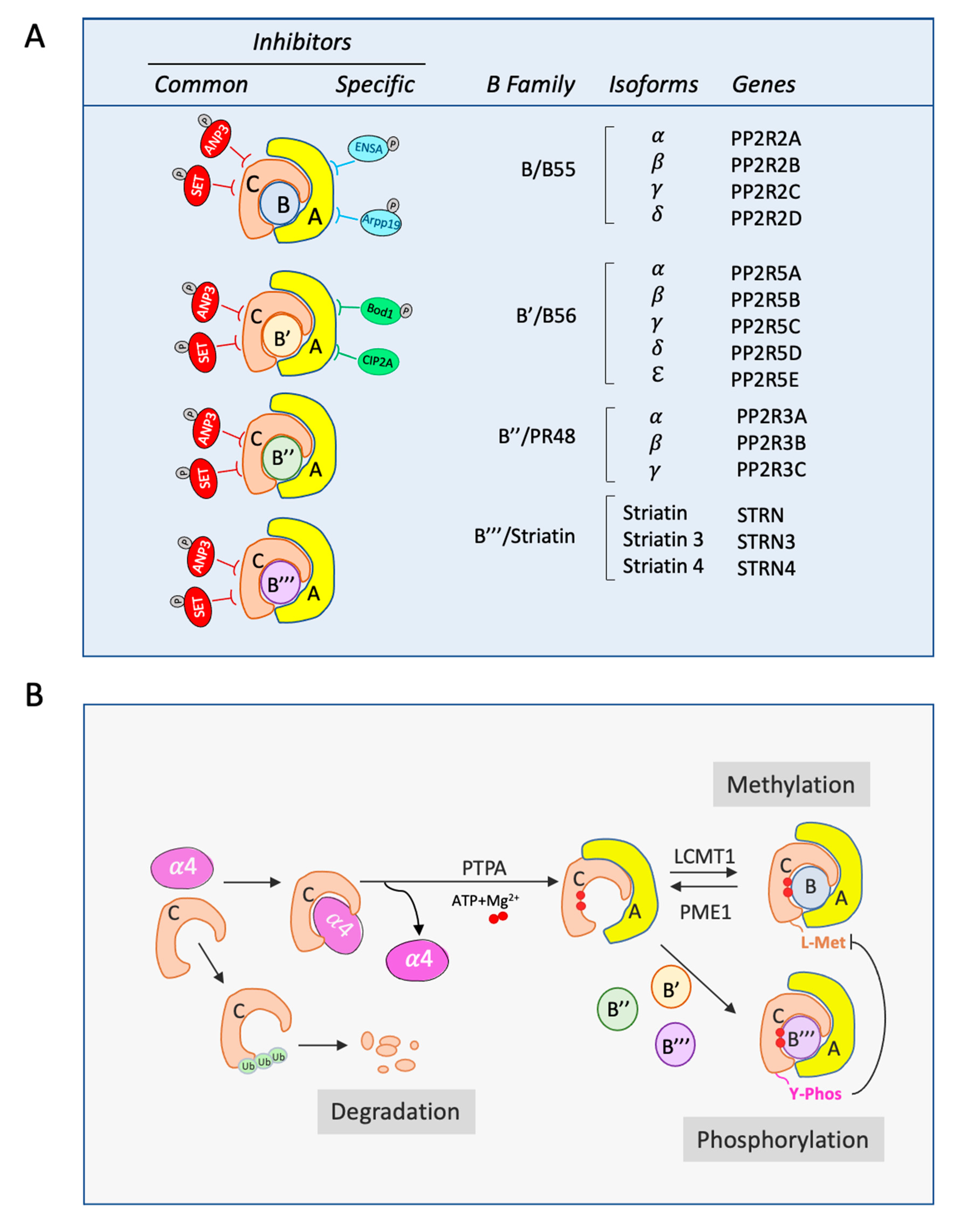
:1. Introduction
2. PP2A Structure
3. PP2A Activation
4. PP2A Endogenous Inhibitors Regulating C Subunit and PP2A-B56
5. PP2A Endogenous Inhibitors Regulating PP2A-B55
6. Signaling Cascades Regulated by PP2A-B55
7. PP2A-B55 Misregulation in Cancer
8. Arpp19 and ENSA Misregulation in Cancer
9. Gwl/Mastl Misregulation in Cancer
10. Summary
Funding
Conflicts of Interest
References
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Lit, L.C.; Zhang, H.; Darrington, R.S.; Melaiu, O.; Rudraraju, B.; Giamas, G. The regulatory roles of phosphatases in cancer. Oncogene 2014, 33, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Wang, Z.; Jiang, C.; Ding, Y. PP2A-Mediated Anticancer Therapy. Gastroenterol. Res. Pract. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Brautigan, D.L.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Hidalgo, L.; Moreno, S. Coupling TOR to the Cell Cycle by the Greatwall-Endosulfine-PP2A-B55 Pathway. Biomolecules 2017, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Crncec, A.; Hochegger, H. Triggering mitosis. FEBS Lett. 2019, 593, 2868–2888. [Google Scholar] [CrossRef] [Green Version]
- Glover, D.M. The overlooked greatwall: A new perspective on mitotic control. Open Biol. 2012, 2, 120023. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.; Lorca, T. Greatwall kinase at a glance. J. Cell Sci. 2018, 131, jcs222364. [Google Scholar] [CrossRef] [Green Version]
- Lorca, T.; Castro, A. The Greatwall kinase: A new pathway in the control of the cell cycle. Oncogene 2013, 32, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorca, T.; Castro, A. Deciphering the New Role of the Greatwall/PP2A Pathway in Cell Cycle Control. Genes Cancer 2012, 3, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, S.; Robert, P.; Hached, K.; Sundermann, L.; Charrasse, S.; Labbe, J.C.; Castro, A.; Lorca, T. The master Greatwall kinase, a critical regulator of mitosis and meiosis. Int. J. Dev. Biol. 2016, 60, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Jessus, C. ARPP19 Phosphorylations by PKA and Greatwall: The Yin and the Yang of the Cell Decision to Divide. Protein Phosphorylation 2017. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv. Biol. Regul. 2013, 53, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Hunt, T. Protein phosphatases and their regulation in the control of mitosis. EMBO Rep. 2012, 13, 197–203. [Google Scholar] [CrossRef]
- Gu, P.; Qi, X.; Zhou, Y.; Wang, Y.; Gao, X. Generation of Ppp2Ca and Ppp2Cb conditional null alleles in mouse. Genesis 2012, 50, 429–436. [Google Scholar] [CrossRef]
- Gotz, J.; Probst, A.; Ehler, E.; Hemmings, B.; Kues, W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit C. Proc. Natl. Acad. Sci. 1998, 95, 12370–12375. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, Y.; Zhang, P.; Jeffrey, P.D.; Shi, Y. Structure of a Protein Phosphatase 2A Holoenzyme: Insights into B55-Mediated Tau Dephosphorylation. Mol. Cell 2008, 31, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Wlodarchak, N.; Guo, F.; Satyshur, K.A.; Jiang, L.; Jeffrey, P.D.; Sun, T.; Stanevich, V.; Mumby, M.C.; Xing, Y. Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res. 2013, 23, 931–946. [Google Scholar] [CrossRef] [Green Version]
- Strack, S.; Chang, D.; Zaucha, J.A.; Colbran, R.J.; Wadzinski, B.E. Cloning and characterization of Bδ, a novel regulatory subunit of protein phosphatase 2A. FEBS Lett. 1999, 460, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Li, Z.; Chen, Y.; Stock, J.B.; Jeffrey, P.D.; Shi, Y. Structural Mechanism of Demethylation and Inactivation of Protein Phosphatase 2A. Cell 2008, 133, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longin, S.; Zwaenepoel, K.; Louis, J.V.; Dilworth, S.; Goris, J.; Janssens, V. Selection of Protein Phosphatase 2A Regulatory Subunits Is Mediated by the C Terminus of the Catalytic Subunit. J. Biol. Chem. 2007, 282, 26971–26980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, S.S.; Perry, J.A.; Forester, C.M.; Nutt, L.K.; Guo, Y.; Jardim, M.J.; Thomenius, M.J.; Freel, C.D.; Darbandi, R.; Ahn, J.-H.; et al. Role for the PP2A/B56δ Phosphatase in Regulating 14-3-3 Release from Cdc25 to Control Mitosis. Cell 2006, 127, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letourneux, C.; Rocher, G.; Porteu, F. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 2006, 25, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, U.S.; Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007, 445, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Low, I.C.C.; Loh, T.; Huang, Y.; Virshup, D.M.; Pervaiz, S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56δ stabilizes its antiapoptotic activity. Blood 2014, 124, 2223–2234. [Google Scholar] [CrossRef]
- Janssens, V.; Jordens, J.; Stevens, I.; Van Hoof, C.; Martens, E.; De Smedt, H.; Engelborghs, Y.; Waelkens, E.; Goris, J. Identification and Functional Analysis of Two Ca 2+ -binding EF-hand Motifs in the B"/PR72 Subunit of Protein Phosphatase 2A. J. Biol. Chem. 2003, 278, 10697–10706. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Fedorov, S.A.; Mumby, M.C.; Williams, R.S. PR48, a Novel Regulatory Subunit of Protein Phosphatase 2A, Interacts with Cdc6 and Modulates DNA Replication in Human Cells. Mol. Cell. Biol. 2000, 20, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Haesen, D.; Sents, W.; Lemaire, K.; Hoorne, Y.; Janssens, V. The Basic Biology of PP2A in Hematologic Cells and Malignancies. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Goudreault, M.; D’Ambrosio, L.M.; Kean, M.J.; Mullin, M.J.; Larsen, B.G.; Sanchez, A.; Chaudhry, S.; Chen, G.I.; Sicheri, F.; Nesvizhskii, A.I.; et al. A PP2A Phosphatase High Density Interaction Network Identifies a Novel Striatin-interacting Phosphatase and Kinase Complex Linked to the Cerebral Cavernous Malformation 3 (CCM3) Protein. Mol. Cell. Proteom. 2009, 8, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.; Ditsworth, D.; Lindsten, T.; Thompson, C.B. α4 is an essential regulator of PP2A phosphatase activity. Mol. Cell 2009, 36, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F. Structural basis of PP2A activation by PTPA, an ATP-dependent activation chaperone. Cell Res. 2014, 24, 190–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.R.H.; Hemmings, B.A. Mutation of the C-terminal leucine residue of PP2Ac inhibits PR55/B subunit binding and confers supersensitivity to microtubule destabilization in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 264, 425–432. [Google Scholar] [CrossRef]
- Wei, H.; Ashby, D.G.; Moreno, C.S.; Ogris, E.; Yeong, F.M.; Corbett, A.H.; Pallas, D.C. Carboxymethylation of the PP2A Catalytic Subunit inSaccharomyces cerevisiae Is Required for Efficient Interaction with the B-type Subunits Cdc55p and Rts1p. J. Biol. Chem. 2001, 276, 1570–1577. [Google Scholar] [CrossRef] [Green Version]
- Gentry, M.S.; Li, Y.; Wei, H.; Syed, F.F.; Patel, S.H.; Hallberg, R.L.; Pallas, D.C. A Novel Assay for Protein Phosphatase 2A (PP2A) Complexes In Vivo Reveals Differential Effects of Covalent Modifications on Different Saccharomyces cerevisiae PP2A Heterotrimers. Eukaryot. Cell 2005, 4, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Koren, R.; Rainis, L.; Kleinberger, T. The Scaffolding A/Tpd3 Subunit and High Phosphatase Activity Are Dispensable for Cdc55 Function in the Saccharomyces cerevisiae Spindle Checkpoint and in Cytokinesis. J. Biol. Chem. 2004, 279, 48598–48606. [Google Scholar] [CrossRef] [Green Version]
- Ogris, E.; Gibson, D.M.; Pallas, D.C. Protein phosphatase 2A subunit assembly: The catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene 1997, 15, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691. [Google Scholar] [CrossRef] [Green Version]
- Nunbhakdi-Craig, V.; Schuechner, S.; Sontag, J.-M.; Montgomery, L.; Pallas, D.C.; Juno, C.; Mudrak, I.; Ogris, E.; Sontag, E. Expression of protein phosphatase 2A mutants and silencing of the regulatory Bα subunit induce a selective loss of acetylated and detyrosinated microtubules. J. Neurochem. 2007, 101, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, H.; Damuni, Z. Purification and Characterization of Two Potent Heat-Stable Protein Inhibitors of Protein Phosphatase 2A from Bovine Kidney. Biochemistry 1995, 34, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Reilly, P.T.; Yu, Y.; Hamiche, A.; Wang, L. Cracking the ANP32 whips: Important functions, unequal requirement, and hints at disease implications. Bioessays 2014, 36, 1062–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. I PP2A 1 Affects Tau Phosphorylation via Association with the Catalytic Subunit of Protein Phosphatase 2A. J. Biol. Chem. 2008, 283, 10513–10521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachr, Y.; Pavlakis, G.N.; Copeland, T.D. Identification of in vivo phosphorylation sites of SET, a nuclear phosphoprotein encoded by the translocation breakpoint in acute undifferentiated leukemia. FEBS Lett. 1994, 340, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Irie, A.; Harada, K.; Araki, N.; Nishimura, Y. Phosphorylation of SET Protein at Ser171 by Protein Kinase D2 Diminishes Its Inhibitory Effect on Protein Phosphatase 2A. PLoS ONE 2012, 7, e51242. [Google Scholar] [CrossRef]
- Vasudevan, N.T.; Mohan, M.L.; Gupta, M.K.; Hussain, A.K.; Prasad, S.V.N. Inhibition of Protein Phosphatase 2A Activity by PI3Kγ Regulates β-Adrenergic Receptor Function. Mol. Cell 2011, 41, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Cristóbal, I.; Blanco, F.J.; Garcia-Orti, L.; Marcotegui, N.; Vicente, C.; Rifon, J.; Novo, F.J.; Bandres, E.; Calasanz, M.J.; Bernabeu, C.; et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood 2010, 115, 615–625. [Google Scholar] [CrossRef]
- Chambon, J.-P.; Touati, S.A.; Berneau, S.; Cladière, D.; Hebras, C.; Groeme, R.; McDougall, A.; Wassmann, K. The PP2A Inhibitor I2PP2A Is Essential for Sister Chromatid Segregation in Oocyte Meiosis II. Curr. Biol. 2013, 23, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A.; et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Leopoldino, A.M.; Squarize, C.H.; Garcia, C.B.; Almeida, L.O.; Pestana, C.R.; Polizello, A.C.M.; Uyemura, S.A.; Tajara, E.H.; Gutkind, J.S.; Curti, C. Accumulation of the SET protein in HEK293T cells and mild oxidative stress: Cell survival or death signaling. Mol. Cell Biochem. 2012, 363, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.J.; Chen, Y.; Oddo, J.; Matta, K.M.; Neil, J.; Davis, E.D.; Volkheimer, A.D.; Lanasa, M.C.; Friedman, D.R.; Goodman, B.K.; et al. SET oncoprotein overexpression in B-cell chronic lymphocytic leukemia and non-Hodgkin lymphoma: A predictor of aggressive disease and a new treatment target. Blood 2011, 118, 4150–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Murrani, S.W.K.; Woodgett, J.R.; Damuni, Z. Expression of I2PP2A, an inhibitor of protein phosphatase 2A, induces c-Jun and AP-1 activity. Biochem. J. 1999, 341, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Chu, P.-Y.; Chen, J.-L.; Huang, C.-T.; Lee, C.-H.; Lau, K.-Y.; Wang, W.-L.; Wang, Y.-L.; Lien, P.-J.; Tseng, L.-M.; et al. SET Overexpression is Associated with Worse Recurrence-Free Survival in Patients with Primary Breast Cancer Receiving Adjuvant Tamoxifen Treatment. J. Clin. Med. 2018, 7, 245. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Gu, Y.; Wang, H.; Yin, J.; Zheng, G.; Zhang, Z.; Lu, M.; Wang, C.; He, Z. Overexpression of PP2A inhibitor SET oncoprotein is associated with tumor progression and poor prognosis in human non-small cell lung cancer. Oncotarget 2015, 6, 14913–14925. [Google Scholar] [CrossRef] [Green Version]
- Brander, D.M.; Friedman, D.R.; Volkheimer, A.D.; Christensen, D.J.; Rassenti, L.Z.; Kipps, T.J.; Guadalupe, E.; Chen, Y.; Zhang, D.; Wang, X.; et al. SET alpha and SET beta mRNA isoforms in chronic lymphocytic leukaemia. Br. J. Haematol. 2019, 184, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Puustinen, P.; Niemelä, M.; Ahola, R.; Arnold, H.; Böttzauw, T.; Ala-aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A Inhibits PP2A in Human Malignancies. Cell 2007, 130, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Okkeri, J.; Pavic, K.; Wang, Z.; Kauko, O.; Halonen, T.; Sarek, G.; Ojala, P.M.; Rao, Z.; Xu, W.; et al. Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep. 2017, 18, 437–450. [Google Scholar] [CrossRef]
- Laine, A.; Sihto, H.; Come, C.; Rosenfeldt, M.T.; Zwolinska, A.; Niemelä, M.; Khanna, A.; Chan, E.K.; Kähäri, V.-M.; Kellokumpu-Lehtinen, P.-L.; et al. Senescence Sensitivity of Breast Cancer Cells Is Defined by Positive Feedback Loop between CIP2A and E2F1. Cancer Discov. 2013, 3, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-F.; Liu, C.-Y.; Lin, Y.-C.; Yu, H.-C.; Liu, T.-H.; Hou, D.-R.; Chen, P.-J.; Cheng, A.-L. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 2010, 29, 6257–6266. [Google Scholar] [CrossRef] [Green Version]
- Khanna, A.; Pimanda, J.E.; Westermarck, J. Cancerous Inhibitor of Protein Phosphatase 2A, an Emerging Human Oncoprotein and a Potential Cancer Therapy Target. Cancer Res. 2013, 73, 6548–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, I.M.; Schleicher, K.; Porter, M.; Swedlow, J.R. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat. Commun. 2013, 4, 2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharbi-Ayachi, A.; Labbe, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef]
- Mochida, S.; Ikeo, S.; Gannon, J.; Hunt, T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009, 28, 2777–2785. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, S.; Brioudes, E.; Burgess, A.; Labbe, J.C.; Lorca, T.; Castro, A. Greatwall maintains mitosis through regulation of PP2A. EMBO J. 2009, 28, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Juanes, M.A.; Khoueiry, R.; Kupka, T.; Castro, A.; Mudrak, I.; Ogris, E.; Lorca, T.; Piatti, S. Budding yeast greatwall and endosulfines control activity and spatial regulation of PP2A(Cdc55) for timely mitotic progression. PLoS Genet. 2013, 9, e1003575. [Google Scholar] [CrossRef] [Green Version]
- Labandera, A.-M.; Vahab, A.R.; Chaudhuri, S.; Kerk, D.; Moorhead, G.B.G. The mitotic PP2A regulator ENSA/ARPP-19 is remarkably conserved across plants and most eukaryotes. Biochem. Biophys. Res. Commun. 2015, 458, 739–744. [Google Scholar] [CrossRef]
- Cundell, M.J.; Bastos, R.N.; Zhang, T.; Holder, J.; Gruneberg, U.; Novak, B.; Barr, F.A. The BEG (PP2A-B55/ENSA/Greatwall) pathway ensures cytokinesis follows chromosome separation. Mol. Cell 2013, 52, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Hached, K.; Goguet, P.; Charrasse, S.; Vigneron, S.; Sacristan, M.P.; Lorca, T.; Castro, A. ENSA and ARPP19 differentially control cell cycle progression and development. J. Cell Biol. 2019, 218, 541–558. [Google Scholar] [CrossRef]
- Hegarat, N.; Vesely, C.; Vinod, P.K.; Ocasio, C.; Peter, N.; Gannon, J.; Oliver, A.W.; Novak, B.; Hochegger, H. PP2A/B55 and Fcp1 regulate Greatwall and Ensa dephosphorylation during mitotic exit. PLoS Genet 2014, 10, e1004004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupré, A.; Daldello, E.M.; Nairn, A.C.; Jessus, C.; Haccard, O. Phosphorylation of ARPP19 by protein kinase A prevents meiosis resumption in Xenopus oocytes. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, L.M.; Evans, J.P. alpha-endosulfine (ENSA) regulates exit from prophase I arrest in mouse oocytes. Cell Cycle 2014, 13, 1639–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrasse, S.; Gharbi-Ayachi, A.; Burgess, A.; Vera, J.; Hached, K.; Raynaud, P.; Schwob, E.; Lorca, T.; Castro, A. Ensa controls S-phase length by modulating Treslin levels. Nat. Commun. 2017, 8, 206. [Google Scholar] [CrossRef]
- Kumm, E.J.; Pagel, O.; Gambaryan, S.; Walter, U.; Zahedi, R.P.; Smolenski, A.; Jurk, K. The Cell Cycle Checkpoint System MAST(L)-ENSA/ARPP19-PP2A is Targeted by cAMP/PKA and cGMP/PKG in Anucleate Human Platelets. Cells 2020, 9, 472. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, M.J.; Cummings, C.L.; Drachman, J.G. FLJ14813 Missense Mutation: A Candidate for Autosomal Dominant Thrombocytopenia on Human Chromosome 10. Hum. Hered. 2003, 55, 66–70. [Google Scholar] [CrossRef]
- Hurtado, B.; Trakala, M.; Ximénez-Embún, P.; El Bakkali, A.; Partida, D.; Sanz-Castillo, B.; Álvarez-Fernández, M.; Maroto, M.; Sánchez-Martínez, R.; Martínez, L.; et al. Thrombocytopenia-associated mutations in Ser/Thr kinase MASTL deregulate actin cytoskeletal dynamics in platelets. J. Clin. Investig. 2018, 128, 5351–5367. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [Green Version]
- Ory, S.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Morrison, D.K. Protein Phosphatase 2A Positively Regulates Ras Signaling by Dephosphorylating KSR1 and Raf-1 on Critical 14-3-3 Binding Sites. Curr. Biol. 2003, 13, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Fritz, A.; Brayer, K.J.; McCormick, N.; Adams, D.G.; Wadzinski, B.E.; Vaillancourt, R.R. Phosphorylation of Serine 526 Is Required for MEKK3 Activity, and Association with 14-3-3 Blocks Dephosphorylation. J. Biol. Chem. 2006, 281, 6236–6245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, P.J.A.; Creyghton, M.P.; Wilhelmsen, K.; van Dam, H.; Bernards, R. A RNA Interference Screen Identifies the Protein Phosphatase 2A Subunit PR55c as a Stress-Sensitive Inhibitor of c-SRC. PLoS Genet 2007, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, J.; Liu, Y.; Chen, X.; Yu, T.; Jia, J.; Liu, C. PR55α, a Regulatory Subunit of PP2A, Specifically Regulates PP2A-mediated β-Catenin Dephosphorylation. J. Biol. Chem. 2009, 284, 22649–22656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.-M.; Feng, Y.; Wang, J.; Shi, R.; Jiang, X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015, 6, 8048. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Di Conza, G.; Cafarello, S.T.; Loroch, S.; Mennerich, D.; Deschoemaeker, S.; Di Matteo, M.; Ehling, M.; Gevaert, K.; Prenen, H.; Zahedi, R.P.; et al. The mTOR and PP2A Pathways Regulate PHD2 Phosphorylation to Fine-Tune HIF1α Levels and Colorectal Cancer Cell Survival under Hypoxia. Cell Rep. 2017, 18, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, H.; Suganuma, M. Tumor Promotion by Inhibitors of ProteinZ Phosphatases 1 and 2A: The Okadaic Acid Class of Compounds. In Advances in Cancer Research; Woude, G.F.V., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 1993; Volume 61, pp. 143–194. [Google Scholar]
- Dilworth, S.M. Polyoma virus middle T antigen and its role in identifying cancer-related molecules. Nat. Rev. Cancer 2002, 2, 951–956. [Google Scholar] [CrossRef]
- Skoczylas, C.; Fahrbach, K.M.; Rundell, K. Cellular targets of the SV40 small-t antigen in human cell transformation. Cell Cycle 2004, 3, 606–610. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, W.; Kim, S.T.; Sun, J.; Lu, L.; Sun, J.; Zheng, S.L.; Isaacs, W.B.; Xu, J. Evaluation of PPP2R2A as a prostate cancer susceptibility gene: A comprehensive germline and somatic study. Cancer Genet. 2011, 204, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Mosca, L.; Musto, P.; Todoerti, K.; Barbieri, M.; Agnelli, L.; Fabris, S.; Tuana, G.; Lionetti, M.; Bonaparte, E.; Sirchia, S.M.; et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am. J. Hematol. 2013, 88, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Sakata-Yanagimoto, M.; Sanada, M.; Sato-Otsubo, A.; Enami, T.; Suzukawa, K.; Kurita, N.; Nishikii, H.; Yokoyama, Y.; Okoshi, Y.; et al. Identification of unbalanced genome copy number abnormalities in patients with multiple myeloma by single-nucleotide polymorphism genotyping microarray analysis. Int. J. Hematol. 2012, 96, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Shouse, G.; de Necochea-Campion, R.; Mirshahidi, S.; Liu, X.; Chen, C.-S. Novel B55α-PP2A mutations in AML promote AKT T308 phosphorylation and sensitivity to AKT inhibitor-induced growth arrest. Oncotarget 2016, 7, 61081–61092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, P.P.; Qui, Y.H.; Coombes, K.R.; Zhang, N.; Ruvolo, V.R.; Borthakur, G.; Konopleva, M.; Andreeff, M.; Kornblau, S.M. Low expression of PP2A regulatory subunit B55α is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia 2011, 25, 1711–1717. [Google Scholar] [CrossRef]
- Kalev, P.; Simicek, M.; Vazquez, I.; Munck, S.; Chen, L.; Soin, T.; Danda, N.; Chen, W.; Sablina, A. Loss of PPP2R2A Inhibits Homologous Recombination DNA Repair and Predicts Tumor Sensitivity to PARP Inhibition. Cancer Res. 2012, 72, 6414–6424. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Lee, P.L.; Li, Z.; Jiang, X.; Lim, Y.C.; Hooi, S.C.; Yu, Q. B55β-Associated PP2A Complex Controls PDK1-Directed Myc Signaling and Modulates Rapamycin Sensitivity in Colorectal Cancer. Cancer Cell 2010, 18, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Muggerud, A.A.; Rønneberg, J.A.; Wärnberg, F.; Botling, J.; Busato, F.; Jovanovic, J.; Solvang, H.; Bukholm, I.; Børresen-Dale, A.-L.; Kristensen, V.N.; et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. 2010, 12, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bluemn, E.G.; Spencer, E.S.; Mecham, B.; Gordon, R.R.; Coleman, I.; Lewinshtein, D.; Mostaghel, E.; Zhang, X.; Annis, J.; Grandori, C.; et al. PPP2R2C Loss Promotes Castration-Resistance and Is Associated with Increased Prostate Cancer-Specific Mortality. Mol. Cancer Res. 2013, 11, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Q.; Zhou, T.; He, C.; Zhang, S.; Qiu, Y.; Luo, B.; Zhao, R.; Liu, H.; Lin, Y.; Lin, Z. Protein phosphatase 2A-B55δ enhances chemotherapy sensitivity of human hepatocellular carcinoma under the regulation of microRNA-133b. J. Exp. Clin. Cancer Res. 2016, 35, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, R.; Makhijani, K.; Rao, P.R.; Shashidhara, L.S. Drosophila Twins regulates Armadillo levels in response to Wg/Wnt signal. Development 2004, 131, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, A.L.; Seshacharyulu, P.; Rachagani, S.; Sheinin, Y.M.; Ouellette, M.M.; Ponnusamy, M.P.; Mumby, M.C.; Batra, S.K.; Yan, Y. PR55α Subunit of Protein Phosphatase 2A Supports the Tumorigenic and Metastatic Potential of Pancreatic Cancer Cells by Sustaining Hyperactive Oncogenic Signaling. Cancer Res. 2016, 76, 2243–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Xie, G.; Sun, Q. Long Noncoding RNA DLX6-AS1 Promotes the Progression in Cervical Cancer by Targeting miR-16-5p/ARPP19 Axis. Cancer Biother. Radiopharm. 2020, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, Y.; Pu, Y.-S.; Cui, M.-L.; Mao, Z.-J.; Li, Z.-Z.; He, L.; Wu, M.; Wang, J.-H. LncRNA IGFL2-AS1 functions as a ceRNA in regulating ARPP19 through competitive binding to miR-802 in gastric cancer. Mol. Carcinog. 2020, 59, 311–322. [Google Scholar] [CrossRef]
- Ye, H.; Jin, Q.; Wang, X.; Li, Y. MicroRNA-802 Inhibits Cell Proliferation and Induces Apoptosis in Human Laryngeal Cancer by Targeting cAMP-Regulated Phosphoprotein 19. Available online: https://www.dovepress.com/microrna-802-inhibits-cell-proliferation-and-induces-apoptosis-in-huma-peer-reviewed-article-CMAR (accessed on 28 May 2020).
- Gong, Y.; Wu, W.; Zou, X.; Liu, F.; Wei, T.; Zhu, J. MiR-26a inhibits thyroid cancer cell proliferation by targeting ARPP19. Am. J. Cancer Res. 2018, 8, 1030–1039. [Google Scholar]
- Lü, M.; Ding, K.; Zhang, G.; Yin, M.; Yao, G.; Tian, H.; Lian, J.; Liu, L.; Liang, M.; Zhu, T.; et al. MicroRNA-320a sensitizes tamoxifen-resistant breast cancer cells to tamoxifen by targeting ARPP-19 and ERRγ*. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Pan, J.; Liu, Y.; Wen, H.; Wang, L.; Cui, J.; Liu, Y.; Hu, B.; Yao, Z.; Ji, G. Increased ARPP-19 Expression Is Associated with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2014, 16, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Mäkelä, E.; Löyttyniemi, E.; Salmenniemi, U.; Kauko, O.; Varila, T.; Kairisto, V.; Itälä-Remes, M.; Westermarck, J. Arpp19 Promotes Myc and Cip2a Expression and Associates with Patient Relapse in Acute Myeloid Leukemia. Cancers 2019, 11, 1774. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-L.; Kuo, M.-H.; Lin, P.-Y.; Chuang, W.-L.; Hsu, C.-C.; Chu, P.-Y.; Lee, C.-H.; Huang, T.H.-M.; Leu, Y.-W.; Hsiao, S.-H. ENSA expression correlates with attenuated tumor propagation in liver cancer. Biochem. Biophys. Res. Commun. 2013, 442, 56–61. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.-J.; Yang, J.-H.; Wang, Y.; Hua, Z.-J.; Liu, D.; Chen, Y.-Q.; Zhang, H.-M.; Zhang, R.; Zhao, J.-S.; et al. Inflammatory cytokine-induced expression of MASTL is involved in hepatocarcinogenesis by regulating cell cycle progression. Oncol. Lett. 2019, 17, 3163–3172. [Google Scholar] [CrossRef]
- Dahlhaus, M.; Burkovski, A.; Hertwig, F.; Mussel, C.; Volland, R.; Fischer, M.; Debatin, K.M.; Kestler, H.A.; Beltinger, C. Boolean modeling identifies Greatwall/MASTL as an important regulator in the AURKA network of neuroblastoma. Cancer Lett. 2016, 371, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luong, V.; Giannini, P.; Peng, A. Mastl kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget 2014, 5, 11479–11489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Fernández, M.; Sanz-Flores, M.; Sanz-Castillo, B.; Salazar-Roa, M.; Partida, D.; Zapatero-Solana, E.; Ali, H.R.; Manchado, E.; Lowe, S.; VanArsdale, T.; et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2018, 25, 828–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.; McCloy, R.A.; Parker, B.L.; Gallego-Ortega, D.; Law, A.M.K.; Chin, V.T.; Conway, J.R.W.; Fey, D.; Millar, E.K.A.; O’Toole, S.; et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 2018, 37, 4518–4533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuge, B.Z.; Du, B.R.; Meng, X.L.; Zhang, Y.Q. MASTL is a potential poor prognostic indicator in ER+ breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2413–2420. [Google Scholar] [PubMed]
- Tian, J.; Lin, Y.; Yu, J. E2F8 confers cisplatin resistance to ER+ breast cancer cells via transcriptionally activating MASTL. Biomed. Pharmacother. 2017, 92, 919–926. [Google Scholar] [CrossRef]
- Sun, X.-J.; Li, Y.-L.; Wang, L.-G.; Liu, L.-Q.; Ma, H.; Hou, W.-H.; Yu, J.-M. Mastl overexpression is associated with epithelial to mesenchymal transition and predicts a poor clinical outcome in gastric cancer. Oncol. Lett. 2017, 14, 7283–7287. [Google Scholar] [CrossRef]
- Uppada, S.B.; Gowrikumar, S.; Ahmad, R.; Kumar, B.; Szeglin, B.; Chen, X.; Smith, J.J.; Batra, S.K.; Singh, A.B.; Dhawan, P. MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/β-catenin signaling. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Vera, J.; Lartigue, L.; Vigneron, S.; Gadea, G.; Gire, V.; Del Rio, M.; Soubeyran, I.; Chibon, F.; Lorca, T.; Castro, A. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. eLife 2015, 4. [Google Scholar] [CrossRef]
- Yoon, Y.N.; Choe, M.H.; Jung, K.-Y.; Hwang, S.-G.; Oh, J.S.; Kim, J.-S. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anania, M.; Gasparri, F.; Cetti, E.; Fraietta, I.; Todoerti, K.; Miranda, C.; Mazzoni, M.; Re, C.; Colombo, R.; Ukmar, G.; et al. Identification of thyroid tumor cell vulnerabilities through a siRNA-based functional screening. Oncotarget 2015, 6, 34629–34648. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Stigter-van Walsum, M.; Buijze, M.; van den Berg, J.; van der Meulen, I.H.; Hodzic, J.; Piersma, S.R.; Pham, T.V.; Jiménez, C.R.; van Beusechem, V.W.; et al. Genome-wide siRNA Screen Identifies the Radiosensitizing Effect of Downregulation of MASTL and FOXM1 in NSCLC. Mol. Cancer 2015, 14, 1434–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cetti, E.; Di Marco, T.; Mauro, G.; Mazzoni, M.; Lecis, D.; Minna, E.; Gioiosa, L.; Brich, S.; Pagliardini, S.; Borrello, M.G.; et al. Mitosis perturbation by MASTL depletion impairs the viability of thyroid tumor cells. Cancer Lett. 2019, 442, 362–372. [Google Scholar] [CrossRef]
- Taskinen, M.E.; Närvä, E.; Conway, J.R.W.; Hinojosa, L.S.; Lilla, S.; Mai, A.; De Franceschi, N.; Elo, L.L.; Grosse, R.; Zanivan, S.; et al. MASTL promotes cell contractility and motility through kinase-independent signaling. J. Cell Biol. 2020, 219, e201906204. [Google Scholar] [CrossRef]


{kind=link}
| PP2A-B55: Tumour Suppressor Activity | ||||
| Subunit | Gene | Alteration | Disease | Ref. |
| B55𝛼 | PPP2R2A | Deletions | Luminal B breast cancer | [91] |
| B55𝛼 | PPP2R2A | Deletions | Prostate cancer | [92] |
| B55𝛼 | PPP2R2A | Deletions | Primary plasma cell leukaemia | [93] |
| B55𝛽 | PPP2R2B | Deletions | Myeloma | [94] |
| B55𝛼 | PPP2R2A | Loss of function mutation | AML | [95] |
| B55𝛼 | PPP2R2A | B55 downregulation | AML | [96] |
| B55𝛼 | PPP2R2A | Decreased mRNA | Lung & thyroid carcinoma | [97] |
| B55𝛽 | PPP2R2B | Epigenetic silencing | Colorectal cancer | [98] |
| B55𝛽 | PPP2R2B | Epigenetic silencing | Ductal breast carcinoma | [99] |
| B55𝛾 | PPP2R2C | B55 downregulation | Prostate cancer | [100] |
| B55𝛿 | PPP2R2D | Up miRNA/down mRNA | Hepatocellular carcinoma | [101] |
| PP2A-B55: Oncogenic Activity | ||||
| Subunit | Gene | Alteration | Disease | Ref. |
| B55𝛼 | PPP2R2A | B55 overexpression | Pancreatic cancer | [103] |
| Arpp19 Overexpression | |||
|---|---|---|---|
| Alteration | miRNA | Disease | Ref. |
| Long Noncoding RNA DLX6-AS1 overexpression | Drop miR-16-5p | Cervical cancer | [104] |
| Long Noncoding RNA IGFL2-AS1 overexpression | Drop miR-802 | Gastric cancer | [105] |
| _ | Drop miR-802 | Laryngeal cancer | [106] |
| _ | Drop miR-26A | Papillary thyroid Cancer | [107] |
| _ | Drop miR-320 | Breast cancer | [108] |
| Increased mRNA levels | _ | Hepatocellular carcinoma | [109] |
| Increased mRNA levels | _ | AML | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goguet-Rubio, P.; Amin, P.; Awal, S.; Vigneron, S.; Charrasse, S.; Mechali, F.; Labbé, J.C.; Lorca, T.; Castro, A. PP2A-B55 Holoenzyme Regulation and Cancer. Biomolecules 2020, 10, 1586. https://doi.org/10.3390/biom10111586
Goguet-Rubio P, Amin P, Awal S, Vigneron S, Charrasse S, Mechali F, Labbé JC, Lorca T, Castro A. PP2A-B55 Holoenzyme Regulation and Cancer. Biomolecules. 2020; 10(11):1586. https://doi.org/10.3390/biom10111586
Chicago/Turabian StyleGoguet-Rubio, Perrine, Priya Amin, Sushil Awal, Suzanne Vigneron, Sophie Charrasse, Francisca Mechali, Jean Claude Labbé, Thierry Lorca, and Anna Castro. 2020. "PP2A-B55 Holoenzyme Regulation and Cancer" Biomolecules 10, no. 11: 1586. https://doi.org/10.3390/biom10111586