HumiR: Web Services, Tools and Databases for Exploring Human microRNA Data


Abstract
:1. Introduction
2. Experiments and Methods
3. Results
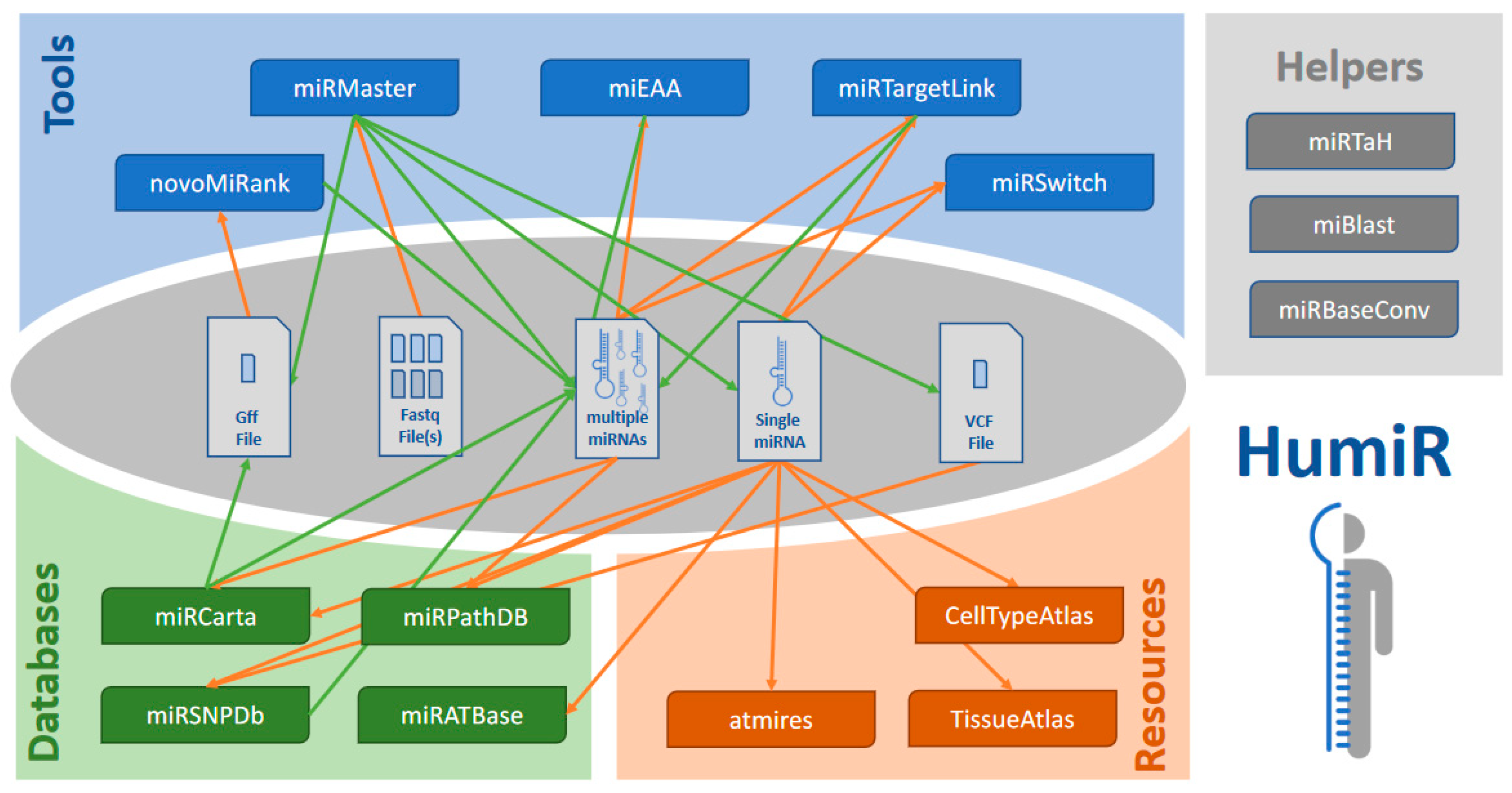
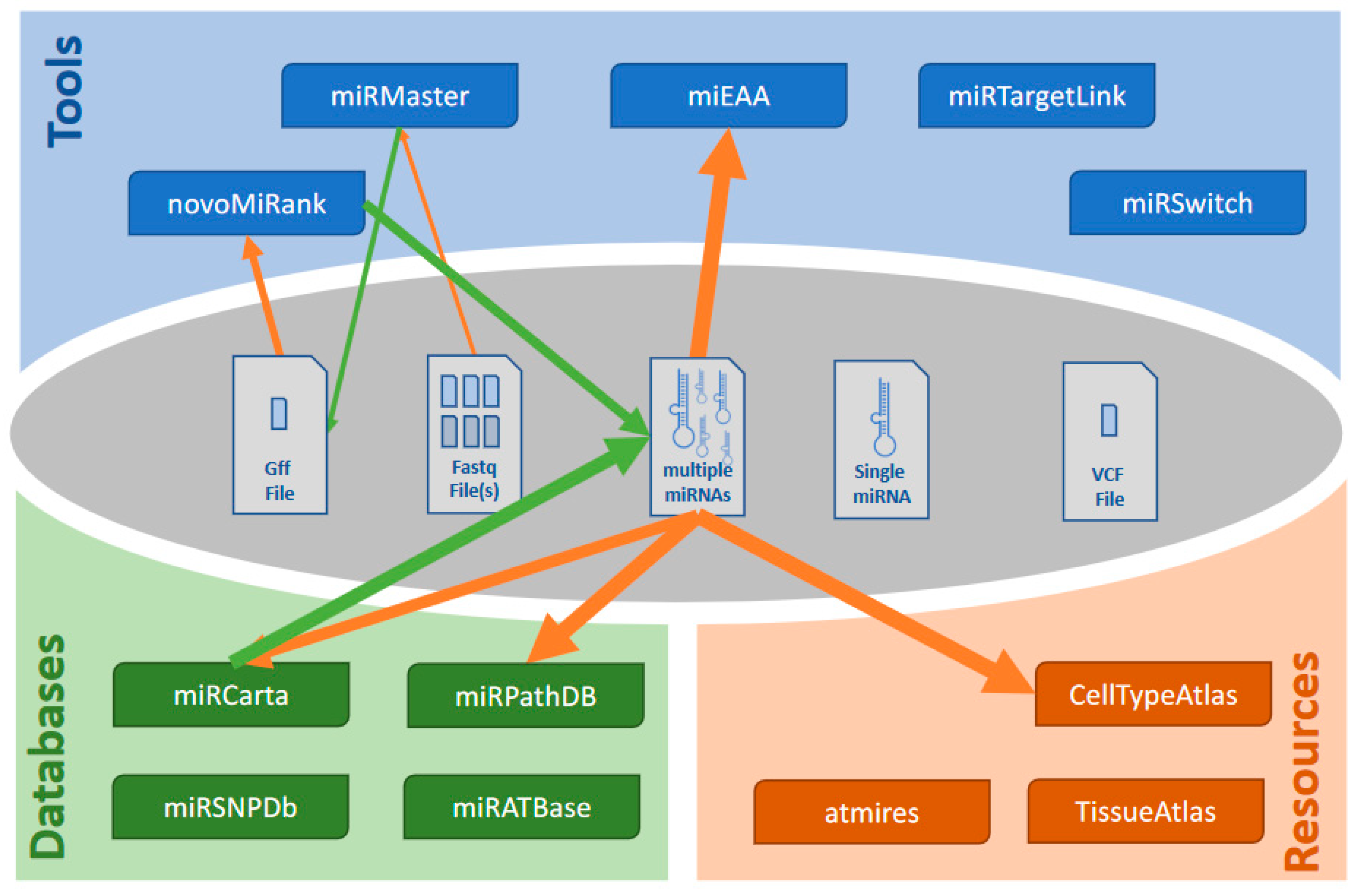
3.1. The Four Categories of Tools
3.2. miRNA Analysis Tools
3.3. Qualitative miRNA Data Bases
3.4. Quantitative miRNA Data Bases
3.5. miRNA Helper Tools
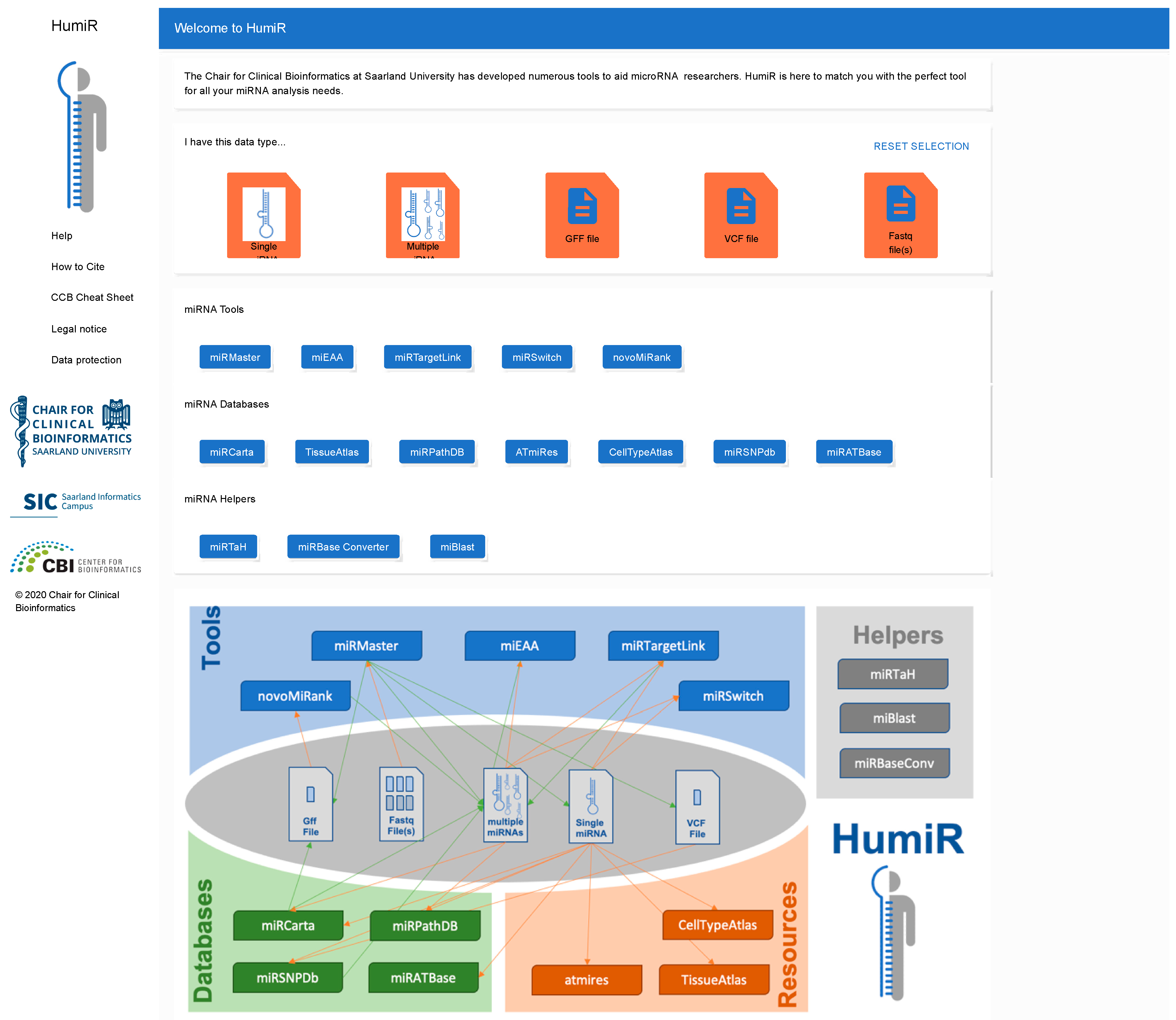
3.6. The HumiR Web Presence

{kind=link}
{kind=link}
{kind=link}
| Category | Tool | Reference | Organism | ncRNA Class | Brief Description/Link |
|---|---|---|---|---|---|
| Analysis | miRMaster | [22,23] | human | miRNA, piRNA, tRNA (+5 others) | non-coding RNA analysis from fastq sequencing files https://www.ccb.uni-saarland.de/mirmaster |
| miEAA | [20,28] | human, mouse, rat (+7 others) | miRNA | miRNA pathway analysis https://www.ccb.uni-saarland.de/mieaa | |
| miRTarget Link | [31] | human | miRNA | analysis of miRNAs and genes in networks https://www.ccb.uni-saarland.de/mirtargetlink | |
| miRSwitch | [34] | human | miRNA | analysis of arm switch and shift events https://www.ccb.uni-saarland.de/mirswitch | |
| NovoMiRank | [35] | human | miRNA | ranking of novel miRNA candidates https://www.ccb.uni-saarland.de/novomirank | |
| Database | miRCarta | [7] | all from miRBase | miRNA | comprehensive collection of miRNAs and miRNA candidates https://www.ccb.uni-saarland.de/mircarta |
| miRSNPdb | [10] | human | miRNA | mutations in human miRNAs https://www.ccb.uni-saarland.de/mirsnp | |
| miRPathDB | [42,43,44] | human, mouse | miRNA | target pathways and categories of miRNAs https://mpd.bioinf.uni-sb.de | |
| miRATbase | NA | human | miRNA | positive and negative reporter assay target validations https://www.ccb.uni-saarland.de/miratbase | |
| TissueAtlas | [47] | human, rat | miRNA | comprehensive atlas of miRNA expression in multiple organs https://www.ccb.uni-saarland.de/tissueatlas | |
| CellTypeAtlas | [48,49] | human | miRNA | comprehensive atlas of miRNA expression in multiple blood cell types https://www.ccb.uni-saarland.de/cf | |
| ATmiRes | [50] | human | miRNA | comprehensive atlas of miRNA expression in ancient samples https://www.ccb.uni-saarland.de/atmires | |
| Helper | miRTaH | NA | all | miRNA | a tool for designing miRNA reporter assay experiments https://www.ccb.uni-saarland.de/mirtah |
| miRBase Converter | [20,28] | all from miRBase | miRNA | converts miRNA identifiers between arbitrary miRBase versions https://www.ccb.uni-saarland.de/mieaa/mirna_version_converter | |
| miBlast | [7] | all from miRBase | miRNA | searches potential novel miRNAs in the miRCarta database https://www.ccb.uni-saarland.de/mircarta/miblast |
3.7. Dependencies and Workflows
4. Outlook: The HumiR Knowledge Base
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [Green Version]
- Lukasik, A.; Wojcikowski, M.; Zielenkiewicz, P. Tools4miRs—One place to gather all the tools for miRNA analysis. Bioinformatics 2016, 32, 2722–2724. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A Uniform System for the Annotation of Vertebrate microRNA Genes and the Evolution of the Human microRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [Green Version]
- Backes, C.; Fehlmann, T.; Kern, F.; Kehl, T.; Lenhof, H.P.; Meese, E.; Keller, A. miRCarta: A central repository for collecting miRNA candidates. Nucleic Acids Res. 2018, 46, D160–D167. [Google Scholar] [CrossRef] [Green Version]
- Fromm, B.; Keller, A.; Yang, X.; Friedlander, M.R.; Peterson, K.J.; Griffiths-Jones, S. Quo vadis microRNAs? Trends Genet 2020, 36, 461–463. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [Green Version]
- Fehlmann, T.; Sahay, S.; Keller, A.; Backes, C. A review of databases predicting the effects of SNPs in miRNA genes or miRNA-binding sites. Brief. Bioinform. 2019, 20, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, X.; Wang, Z.; Li, L.; Chen, H.; Lin, X.; Yi, S.; Zhang, Y.; Xu, J. Systematic review of computational methods for identifying miRNA-mediated RNA-RNA crosstalk. Brief. Bioinform. 2019, 20, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Mrozek, D. A review of Cloud computing technologies for comprehensive microRNA analyses. Comput. Biol. Chem. 2020, 88, 107365. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Backes, C.; Hirsch, P.; Fehlmann, T.; Hart, M.; Meese, E.; Keller, A. What’s the target: Understanding two decades of in silico microRNA-target prediction. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- Stegmayer, G.; Di Persia, L.E.; Rubiolo, M.; Gerard, M.; Pividori, M.; Yones, C.; Bugnon, L.A.; Rodriguez, T.; Raad, J.; Milone, D.H. Predicting novel microRNA: A comprehensive comparison of machine learning approaches. Brief. Bioinform. 2019, 20, 1607–1620. [Google Scholar] [CrossRef] [Green Version]
- Bortolomeazzi, M.; Gaffo, E.; Bortoluzzi, S. A survey of software tools for microRNA discovery and characterization using RNA-seq. Brief. Bioinform. 2019, 20, 918–930. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Kurgan, L. Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief. Bioinform. 2015, 16, 780–794. [Google Scholar] [CrossRef]
- Garcia, A.; Dunoyer-Geindre, S.; Fish, R.J.; Neerman-Arbez, M.; Reny, J.L.; Fontana, P. Methods to Investigate miRNA Function: Focus on Platelet Reactivity. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Le, T.D.; Zhang, J.; Liu, L.; Li, J. Computational methods for identifying miRNA sponge interactions. Brief. Bioinform. 2017, 18, 577–590. [Google Scholar] [CrossRef]
- Kern, F.; Fehlmann, T.; Solomon, J.; Schwed, L.; Grammes, N.; Backes, C.; Van Keuren-Jensen, K.; Craig, D.W.; Meese, E.; Keller, A. miEAA 2.0: Integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res. 2020, 48, W521–W528. [Google Scholar] [CrossRef]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef] [Green Version]
- Fehlmann, T.; Backes, C.; Kahraman, M.; Haas, J.; Ludwig, N.; Posch, A.E.; Wurstle, M.L.; Hubenthal, M.; Franke, A.; Meder, B.; et al. Web-based NGS data analysis using miRMaster: A large-scale meta-analysis of human miRNAs. Nucleic Acids Res. 2017, 45, 8731–8744. [Google Scholar] [CrossRef] [PubMed]
- Fehlmann, T.; Meese, E.; Keller, A. Exploring ncRNAs in Alzheimer’s disease by miRMaster. Oncotarget 2017, 8, 3771–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meistertzheim, M.; Fehlmann, T.; Drews, F.; Pirritano, M.; Gasparoni, G.; Keller, A.; Simon, M. Comparative Analysis of Biochemical Biases by Ligation- and Template-Switch-Based Small RNA Library Preparation Protocols. Clin. Chem. 2019, 65, 1581–1591. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Lebron, R.; Rueda, A.; Gomez-Martin, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. sRNAbench and sRNAtoolbox 2019: Intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Gao, J.; Zhang, H.; Jiang, X.; Zang, Q.; Ban, R.; Zhang, Y.; Shi, Q. CPSS 2.0: A computational platform update for the analysis of small RNA sequencing data. Bioinformatics 2017, 33, 3289–3291. [Google Scholar] [CrossRef]
- Kesharwani, R.K.; Chiesa, M.; Bellazzi, R.; Colombo, G.I. CBS-miRSeq: A comprehensive tool for accurate and extensive analyses of microRNA-sequencing data. Comput. Biol. Med. 2019, 110, 234–243. [Google Scholar] [CrossRef]
- Backes, C.; Khaleeq, Q.T.; Meese, E.; Keller, A. miEAA: MicroRNA enrichment analysis and annotation. Nucleic Acids Res. 2016, 44, W110–W116. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.; Backes, C.; Lenhof, H.P. Computation of significance scores of unweighted Gene Set Enrichment Analyses. BMC Bioinform. 2007, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Hamberg, M.; Backes, C.; Fehlmann, T.; Hart, M.; Meder, B.; Meese, E.; Keller, A. MiRTargetLink—miRNAs, Genes and Interaction Networks. Int. J. Mol. Sci. 2016, 17, 564. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Giroux, P.; Bhajun, R.; Segard, S.; Picquenot, C.; Charavay, C.; Desquilles, L.; Pinna, G.; Ginestier, C.; Denis, J.; Cherradi, N.; et al. miRViz: A novel webserver application to visualize and interpret microRNA datasets. Nucleic Acids Res. 2020, 48, W252–W261. [Google Scholar] [CrossRef] [Green Version]
- Kern, F.; Amand, J.; Senatorov, I.; Isakova, A.; Backes, C.; Meese, E.; Keller, A.; Fehlmann, T. miRSwitch: Detecting microRNA arm shift and switch events. Nucleic Acids Res. 2020, 48, W268–W274. [Google Scholar] [CrossRef]
- Backes, C.; Meder, B.; Hart, M.; Ludwig, N.; Leidinger, P.; Vogel, B.; Galata, V.; Roth, P.; Menegatti, J.; Grasser, F.; et al. Prioritizing and selecting likely novel miRNAs from NGS data. Nucleic Acids Res. 2016, 44, e53. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Becker, M.; Schumann, T.; Speer, T.; Fehlmann, T.; Keller, A.; Meese, E. Bias in recent miRBase annotations potentially associated with RNA quality issues. Sci. Rep. 2017, 7, 5162. [Google Scholar] [CrossRef]
- Backes, C.; Sedaghat-Hamedani, F.; Frese, K.; Hart, M.; Ludwig, N.; Meder, B.; Meese, E.; Keller, A. Bias in High-Throughput Analysis of miRNAs and Implications for Biomarker Studies. Anal. Chem. 2016, 88, 2088–2095. [Google Scholar] [CrossRef]
- Fromm, B.; Domanska, D.; Hoye, E.; Ovchinnikov, V.; Kang, W.; Aparicio-Puerta, E.; Johansen, M.; Flatmark, K.; Mathelier, A.; Hovig, E.; et al. MirGeneDB 2.0: The metazoan microRNA complement. Nucleic Acids Res. 2020, 48, D1172. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhang, F.; Li, T.; Lu, M.; Wang, L.; Yue, W.; Zhang, D. MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genom. 2012, 13, 661. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [PubMed]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stockel, D.; Meese, E.; Lenhof, H.P.; Keller, A. miRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Kehl, T.; Stockel, D.; Fehlmann, T.; Schneider, L.; Meese, E.; Lenhof, H.P.; Keller, A. miRPathDB: A new dictionary on microRNAs and target pathways. Nucleic Acids Res. 2017, 45, D90–D96. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Meese, E.; Lenhof, H.P.; Keller, A. A dictionary on microRNAs and their putative target pathways. Nucleic Acids Res. 2010, 38, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Gerstner, N.; Kehl, T.; Lenhof, K.; Muller, A.; Mayer, C.; Eckhart, L.; Grammes, N.L.; Diener, C.; Hart, M.; Hahn, O.; et al. GeneTrail 3: Advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020, 48, W515–W520. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stahler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Juzenas, S.; Venkatesh, G.; Hubenthal, M.; Hoeppner, M.P.; Du, Z.G.; Paulsen, M.; Rosenstiel, P.; Senger, P.; Hofmann-Apitius, M.; Keller, A.; et al. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 2017, 45, 9290–9301. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, E.C.; Backes, C.; Knorck, A.; Ludwig, N.; Leidinger, P.; Hoxha, C.; Schwar, G.; Grossmann, T.; Muller, S.C.; Hart, M.; et al. Deep characterization of blood cell miRNomes by NGS. Cell. Mol. Life Sci. 2016, 73, 3169–3181. [Google Scholar] [CrossRef]
- Keller, A.; Kreis, S.; Leidinger, P.; Maixner, F.; Ludwig, N.; Backes, C.; Galata, V.; Guerriero, G.; Fehlmann, T.; Franke, A.; et al. miRNAs in Ancient Tissue Specimens of the Tyrolean Iceman. Mol. Biol. Evol. 2017, 34, 793–801. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solomon, J.; Kern, F.; Fehlmann, T.; Meese, E.; Keller, A. HumiR: Web Services, Tools and Databases for Exploring Human microRNA Data. Biomolecules 2020, 10, 1576. https://doi.org/10.3390/biom10111576
Solomon J, Kern F, Fehlmann T, Meese E, Keller A. HumiR: Web Services, Tools and Databases for Exploring Human microRNA Data. Biomolecules. 2020; 10(11):1576. https://doi.org/10.3390/biom10111576
Chicago/Turabian StyleSolomon, Jeffrey, Fabian Kern, Tobias Fehlmann, Eckart Meese, and Andreas Keller. 2020. "HumiR: Web Services, Tools and Databases for Exploring Human microRNA Data" Biomolecules 10, no. 11: 1576. https://doi.org/10.3390/biom10111576