Sphingomyelinases and Liver Diseases

 ,
,
Abstract
:1. Introduction: Metabolism and Regulation of Sphingolipids
2. Ceramide Generation: De Novo Synthesis and Sphingomyelin Hydrolysis by Sphingomyelinases
2.1. De Novo Synthesis
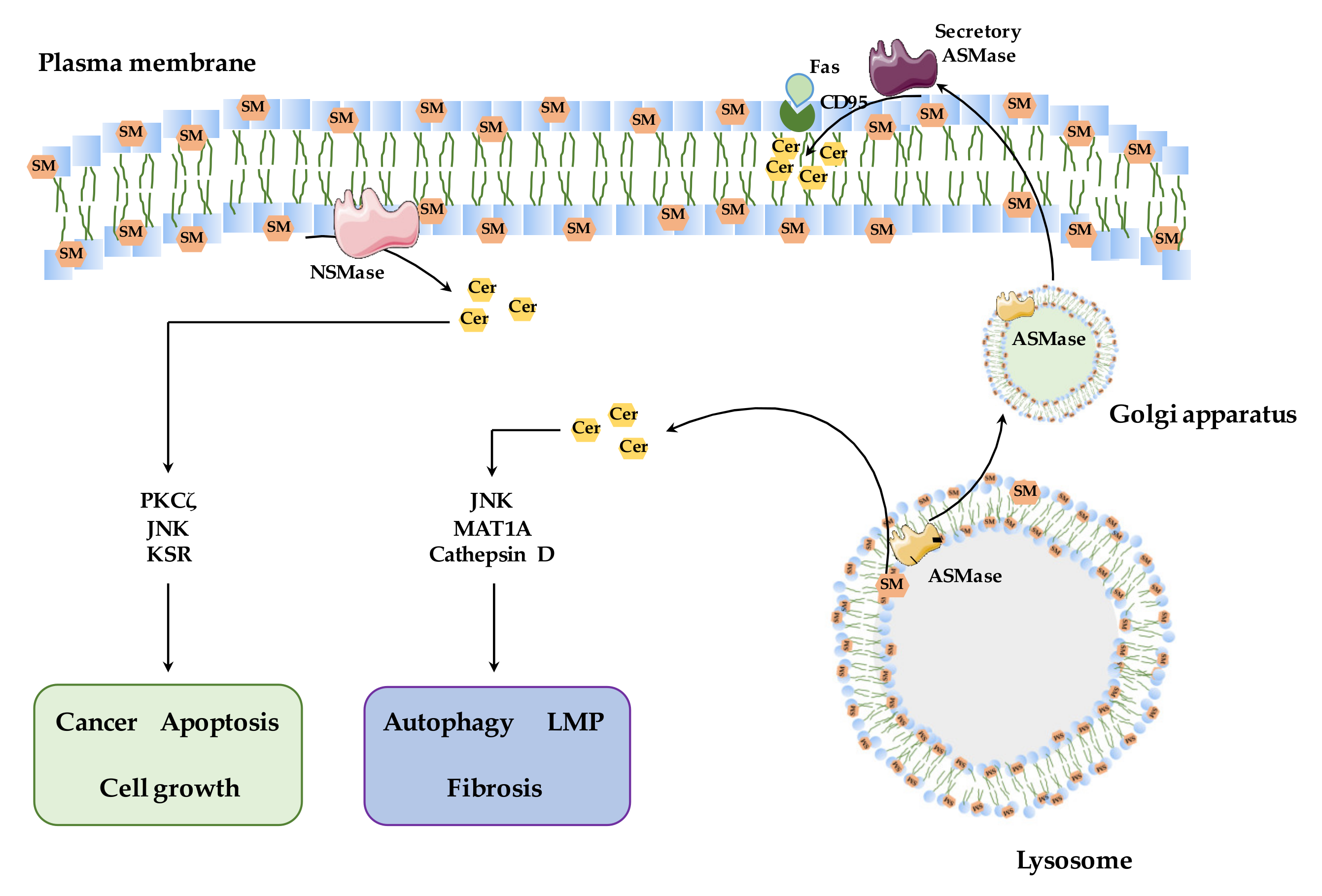
2.2. Sphingomyelinases: Types and Function
3. Physiological and Signaling Function of Sphingomyelinases
3.1. NSMase
3.2. ASMase
3.2.1. ASMase and ER Stress
3.2.2. ASMase and Autophagy
3.2.3. ASMase and Lysosomal Membrane Permeabilization
4. SMases and Liver Diseases
4.1. Alcoholic and Non-Alcoholic Steatohepatitis
4.2. Hepatocellular Carcinoma (HCC)
4.3. Niemann–Pick Disease Type A/B
5. Role of SMases in Liver Injury and Metabolic Liver Diseases
5.1. Ischemia–Reperfusion (I/R) Liver Injury
5.2. Drug-Induced Liver Injury (DILI)
5.3. Viral Hepatitis B (HBV)
5.4. Hepatobiliary Diseases
5.5. Wilson Disease
6. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nikolova-Karakashian, M. Alcoholic and non-alcoholic fatty liver disease: Focus on ceramide. Adv. Biol. Regul. 2018, 70, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Colacios, C.; Sabourdy, F.; Andrieu-Abadie, N.; Ségui, B.; Levade, T. Basics of Sphingolipid Metabolism and Signalling. In Bioactive Sphingolipids in Cancer Biology and Therapy; Hannun, Y.A., Luberto, C., Mao, C., Obeid, L.M., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–20. ISBN 978-3-319-20750-6. [Google Scholar]
- Hannun, Y.A.; Luberto, C.; Mao, C.; Obeid, L.M. Bioactive Sphingolipids in Cancer Biology and Therapy; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. Lond. Engl. 2010, 6, 1603–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschberg, K.; Rodger, J.; Futerman, A.H. The long-chain sphingoid base of sphingolipids is acylated at the cytosolic surface of the endoplasmic reticulum in rat liver. Biochem. J. 1993, 290, 751–757. [Google Scholar] [CrossRef]
- Jaffrézou, J.P.; Levade, T.; Bettaïeb, A.; Andrieu, N.; Bezombes, C.; Maestre, N.; Vermeersch, S.; Rousse, A.; Laurent, G. Daunorubicin-induced apoptosis: Triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 1996, 15, 2417–2424. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Merrill, A.H. Sphingolipid metabolism and cell growth regulation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 1388–1397. [Google Scholar] [CrossRef]
- Hannun, Y.A. The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 1994, 269, 3125–3128. [Google Scholar]
- Kolesnick, R.; Golde, D.W. The sphingomyelin pathway in tumor necrosis factor and interleukin-1 signaling. Cell 1994, 77, 325–328. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Krönke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kihara, A.; Chiba, H.; Tojo, H.; Igarashi, Y. 2-Hydroxy-ceramide synthesis by ceramide synthase family: Enzymatic basis for the preference of FA chain length. J. Lipid Res. 2008, 49, 2356–2364. [Google Scholar] [CrossRef] [Green Version]
- Grösch, S.; Schiffmann, S.; Geisslinger, G. Chain length-specific properties of ceramides. Prog. Lipid Res. 2012, 51, 50–62. [Google Scholar] [CrossRef]
- Pewzner-Jung, Y.; Park, H.; Laviad, E.L.; Silva, L.C.; Lahiri, S.; Stiban, J.; Erez-Roman, R.; Brügger, B.; Sachsenheimer, T.; Wieland, F.; et al. A Critical Role for Ceramide Synthase 2 in Liver Homeostasis. J. Biol. Chem. 2010, 285, 10902–10910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pewzner-Jung, Y.; Brenner, O.; Braun, S.; Laviad, E.L.; Ben-Dor, S.; Feldmesser, E.; Horn-Saban, S.; Amann-Zalcenstein, D.; Raanan, C.; Berkutzki, T.; et al. A critical role for ceramide synthase 2 in liver homeostasis: II. Insights into molecular changes leading to hepatopathy. J. Biol. Chem. 2010, 285, 10911–10923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, D.K.; Weissman, J.S. Membranes in balance: Mechanisms of sphingolipid homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schissel, S.L.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J. Biol. Chem. 1996, 271, 18431–18436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schissel, S.L.; Keesler, G.A.; Schuchman, E.H.; Williams, K.J.; Tabas, I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J. Biol. Chem. 1998, 273, 18250–18259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Canals, D.; Idkowiak-Baldys, J.; Simbari, F.; Roddy, P.; Perry, D.M.; Kitatani, K.; Luberto, C.; Hannun, Y.A. Regulated secretion of acid sphingomyelinase: Implications for selectivity of ceramide formation. J. Biol. Chem. 2010, 285, 35706–35718. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, A.; Illes, K.; Heinz, L.X.; Superti-Furga, G.; Nagar, B. Crystal structure of mammalian acid sphingomyelinase. Nat. Commun. 2016, 7, 12196. [Google Scholar] [CrossRef]
- Andrews, N.W. Solving the secretory acid sphingomyelinase puzzle: Insights from lysosome-mediated parasite invasion and plasma membrane repair. Cell. Microbiol. 2019, 21, e13065. [Google Scholar] [CrossRef]
- Airola, M.V.; Hannun, Y.A. Sphingolipid metabolism and neutral sphingomyelinases. Handb. Exp. Pharmacol. 2013, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Tomiuk, S.; Zumbansen, M.; Stoffel, W. Characterization and subcellular localization of murine and human magnesium-dependent neutral sphingomyelinase. J. Biol. Chem. 2000, 275, 5710–5717. [Google Scholar] [CrossRef] [Green Version]
- Neuberger, Y.; Shogomori, H.; Levy, Z.; Fainzilber, M.; Futerman, A.H. A lyso-platelet activating factor phospholipase C, originally suggested to be a neutral-sphingomyelinase, is located in the endoplasmic reticulum. FEBS Lett. 2000, 469, 44–46. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Lima, F.; Fensome, A.C.; Josephs, M.; Evans, J.; Veldman, R.J.; Katan, M. Structural requirements for catalysis and membrane targeting of mammalian enzymes with neutral sphingomyelinase and lysophospholipid phospholipase C activities. Analysis by chemical modification and site-directed mutagenesis. J. Biol. Chem. 2000, 275, 28316–28325. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, N.; Osta, W.; Bielawski, J.; Luberto, C.; Obeid, L.M.; Hannun, Y.A. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J. Biol. Chem. 2004, 279, 25101–25111. [Google Scholar] [CrossRef] [Green Version]
- Karakashian, A.A.; Giltiay, N.V.; Smith, G.M.; Nikolova-Karakashian, M.N. Expression of neutral sphingomyelinase-2 (NSMase-2) in primary rat hepatocytes modulates IL-beta-induced JNK activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 968–970. [Google Scholar] [CrossRef] [Green Version]
- De Palma, C.; Meacci, E.; Perrotta, C.; Bruni, P.; Clementi, E. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: A novel pathway relevant to the pathophysiology of endothelium. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiuk, S.; Hofmann, K.; Nix, M.; Zumbansen, M.; Stoffel, W. Cloned mammalian neutral sphingomyelinase: Functions in sphingolipid signaling? Proc. Natl. Acad. Sci. USA 1998, 95, 3638–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tepper, A.D.; Ruurs, P.; Borst, J.; van Blitterswijk, W.J. Effect of overexpression of a neutral sphingomyelinase on CD95-induced ceramide production and apoptosis. Biochem. Biophys. Res. Commun. 2001, 280, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.X.; Rajagopalan, V.; Roddy, P.L.; Clarke, C.J.; Hannun, Y.A. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J. Biol. Chem. 2010, 285, 17993–18002. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Nilsson, A.; Tömquist, E.; Duan, R.D. Purification, characterization, and expression of rat intestinal alkaline sphingomyelinase. J. Lipid Res. 2002, 43, 316–324. [Google Scholar]
- Duan, R.-D.; Cheng, Y.; Hansen, G.; Hertervig, E.; Liu, J.-J.; Syk, I.; Sjostrom, H.; Nilsson, A. Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J. Lipid Res. 2003, 44, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.-D. Alkaline sphingomyelinase (NPP7) in hepatobiliary diseases: A field that needs to be closely studied. World J. Hepatol. 2018, 10, 246–253. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Filosto, S.; Fry, W.; Knowlton, A.A.; Goldkorn, T. Neutral sphingomyelinase 2 (nSMase2) is a phosphoprotein regulated by calcineurin (PP2B). J. Biol. Chem. 2010, 285, 10213–10222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, D.A.; Kilkus, J.P.; Gottschalk, A.R.; Quintáns, J.; Dawson, G. Anti-immunoglobulin-induced apoptosis in WEHI 231 cells involves the slow formation of ceramide from sphingomyelin and is blocked by bcl-XL. J. Biol. Chem. 1997, 272, 9868–9876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Kaplowitz, N.; Than, T.A.; Shinohara, M.; Ji, C. Endoplasmic reticulum stress and liver injury. Semin. Liver Dis. 2007, 27, 367–377. [Google Scholar] [CrossRef]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Czaja, M.J.; Ding, W.-X.; Donohue, T.M.; Friedman, S.L.; Kim, J.-S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.J.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Kraft, C.; Peter, M.; Hofmann, K. Selective autophagy: Ubiquitin-mediated recognition and beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Fucho, R.; Martínez, L.; Baulies, A.; Torres, S.; Tarrats, N.; Fernandez, A.; Ribas, V.; Astudillo, A.M.; Balsinde, J.; Garcia-Rovés, P.; et al. Asmase Regulates Autophagy and Lysosomal Membrane Permeabilization and its Inhibition Prevents Early Stage Nonalcoholic Steatohepatitis. J. Hepatol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsin B overexpression due to Acid sphingomyelinase ablation promotes liver fibrosis in niemann-pick disease. J. Biol. Chem. 2012, 287, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Fernandez-Checa, J.C. Damage Mediated by Dysfunction of Organelles and Cellular Systems: Lysosomes. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 97–107. ISBN 978-0-12-386457-4. [Google Scholar]
- Brunk, U.T.; Dalen, H.; Roberg, K.; Hellquist, H.B. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic. Biol. Med. 1997, 23, 616–626. [Google Scholar] [CrossRef]
- Gursahani, H.I.; Schaefer, S. Acidification reduces mitochondrial calcium uptake in rat cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2659–H2665. [Google Scholar] [CrossRef]
- Nilsson, C.; Johansson, U.; Johansson, A.-C.; Kågedal, K.; Ollinger, K. Cytosolic acidification and lysosomal alkalinization during TNF-alpha induced apoptosis in U937 cells. Apoptosis 2006, 11, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egnatchik, R.A.; Leamy, A.K.; Jacobson, D.A.; Shiota, M.; Young, J.D. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol. Metab. 2014, 3, 544–553. [Google Scholar] [CrossRef]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepää, A.-L.; Yki-Järvinen, H.; Oresic, M. Comparison of lipid and fatty acid composition of the liver, subcutaneous and intra-abdominal adipose tissue, and serum. Obesity 2010, 18, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Le Kim, D.; Betzing, H. Intestinal absorption of polyunsaturated phosphatidylcholine in the rat. Hoppe Seylers Z. Physiol. Chem. 1976, 357, 1321–1331. [Google Scholar] [CrossRef]
- Nilsson, Å. Metabolism of sphingomyelin in the intestinal tract of the rat. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1968, 164, 575–584. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [CrossRef]
- Choi, S.; Snider, A.J. Sphingolipids in High Fat Diet and Obesity-Related Diseases. Mediat. Inflamm. 2015, 2015, 520618. [Google Scholar] [CrossRef] [Green Version]
- Memon, R.A.; Holleran, W.M.; Moser, A.H.; Seki, T.; Uchida, Y.; Fuller, J.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-F.; Qu, F.; Zheng, S.-J.; Wu, H.-L.; Liu, M.; Liu, S.; Ren, Y.; Ren, F.; Chen, Y.; Duan, Z.-P.; et al. Elevated plasma sphingomyelin (d18:1/22:0) is closely related to hepatic steatosis in patients with chronic hepatitis C virus infection. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2014, 33, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Koumanov, K.; Infante, R. Phospholipid-transfer proteins in human liver and primary liver carcinoma. Biochim. Biophys. Acta 1986, 876, 526–532. [Google Scholar] [CrossRef]
- Zeidan, Y.H.; Hannun, Y.A. The acid sphingomyelinase/ceramide pathway: Biomedical significance and mechanisms of regulation. Curr. Mol. Med. 2010, 10, 454–466. [Google Scholar] [CrossRef]
- Tilg, H.; Diehl, A.M. Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med. 2000, 343, 1467–1476. [Google Scholar] [CrossRef]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and necrosis in the liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [CrossRef] [Green Version]
- Leake, I. ASMase implicated in alcoholic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 384–385. [Google Scholar] [CrossRef]
- Deaciuc, I.V.; Nikolova-Karakashian, M.; Fortunato, F.; Lee, E.Y.; Hill, D.B.; McClain, C.J. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol. Clin. Exp. Res. 2000, 24, 1557–1565. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Rahmini, Y.; Ross, R.A.; Zhao, Z.; Xu, Y.; Crabb, D.W. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G515–G523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühle, C.; Weinland, C.; Gulbins, E.; Lenz, B.; Kornhuber, J. Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls. Int. J. Mol. Sci. 2018, 19, 4028. [Google Scholar] [CrossRef] [Green Version]
- Grammatikos, G.; Mühle, C.; Ferreiros, N.; Schroeter, S.; Bogdanou, D.; Schwalm, S.; Hintereder, G.; Kornhuber, J.; Zeuzem, S.; Sarrazin, C.; et al. Serum acid sphingomyelinase is upregulated in chronic hepatitis C infection and non alcoholic fatty liver disease. Biochim. Biophys. Acta 2014, 1841, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Tarrats, N.; Morales, A.; Domínguez, M.; Bataller, R.; Caballería, J.; García-Ruiz, C.; Fernández-Checa, J.C.; Marí, M. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am. J. Pathol. 2010, 177, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Colell, A.; Morales, A.; Pañeda, C.; Varela-Nieto, I.; García-Ruiz, C.; Fernández-Checa, J.C. Acidic sphingomyelinase downregulates the liver-specific methionine adenosyltransferase 1A, contributing to tumor necrosis factor-induced lethal hepatitis. J. Clin. Investig. 2004, 113, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Genestier, L.; Pinkoski, M.J.; Castro, A.; Nicholas, S.; Mogil, R.; Paris, F.; Fuks, Z.; Schuchman, E.H.; Kolesnick, R.N.; et al. Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J. Biol. Chem. 2000, 275, 8657–8663. [Google Scholar] [CrossRef] [Green Version]
- Colell, A.; García-Ruiz, C.; Roman, J.; Ballesta, A.; FernándezCheca, J.C. Ganglioside GD3 enhances apoptosis by suppressing the nuclear factor-κB-dependent survival pathway. FASEB J. 2001, 15, 1068–1070. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J. Biol. Chem. 2002, 277, 36443–36448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quillin, R.C.; Wilson, G.C.; Nojima, H.; Freeman, C.M.; Wang, J.; Schuster, R.M.; Blanchard, J.A.; Edwards, M.J.; Gandhi, C.R.; Gulbins, E.; et al. Inhibition of acidic sphingomyelinase reduces established hepatic fibrosis in mice. Hepatol. Res. 2015, 45, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic Reticulum Stress Induces Fibrogenic Activity in Hepatic Stellate Cells through Autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Friedman, S.L. Autophagy fuels tissue fibrogenesis. Autophagy 2012, 8, 849–850. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annu. Rev. Nutr. 2008, 28, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Martin, P.G.P.; Levade, T.; Benoist, H.; Ségui, B. FAN (factor associated with neutral sphingomyelinase activation), a moonlighting protein in TNF-R1 signaling. J. Leukoc. Biol. 2010, 88, 897–903. [Google Scholar] [CrossRef]
- Ségui, B.; Cuvillier, O.; Adam-Klages, S.; Garcia, V.; Malagarie-Cazenave, S.; Lévêque, S.; Caspar-Bauguil, S.; Coudert, J.; Salvayre, R.; Krönke, M.; et al. Involvement of FAN in TNF-induced apoptosis. J. Clin. Investig. 2001, 108, 143–151. [Google Scholar] [CrossRef]
- Krautbauer, S.; Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Weiss, T.S.; Sigruener, A.; Aslanidis, C.; Liebisch, G.; Buechler, C. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2016, 1861, 1767–1774. [Google Scholar] [CrossRef]
- Maeng, H.J.; Song, J.-H.; Kim, G.-T.; Song, Y.-J.; Lee, K.; Kim, J.-Y.; Park, T.-S. Celecoxib-mediated activation of endoplasmic reticulum stress induces de novo ceramide biosynthesis and apoptosis in hepatoma HepG2 cells. BMB Rep. 2017, 50, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, A.; Mao, C.; Jenkins, R.W.; Szulc, Z.M.; Bielawska, A.; Hannun, Y.A. Anticancer actions of lysosomally targeted inhibitor, LCL521, of acid ceramidase. PLoS ONE 2017, 12, e0177805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Liao, W.; Dong, M.; Zhu, R.; Xiao, J.; Sun, T.; Chen, Z.; Wu, B.; Jin, J. Exosomal neutral sphingomyelinase 1 suppresses hepatocellular carcinoma via decreasing the ratio of sphingomyelin/ceramide. FEBS J. 2018, 285, 3835–3848. [Google Scholar] [CrossRef]
- Zhong, L.; Kong, J.N.; Dinkins, M.B.; Leanhart, S.; Zhu, Z.; Spassieva, S.D.; Qin, H.; Lin, H.-P.; Elsherbini, A.; Wang, R.; et al. Increased liver tumor formation in neutral sphingomyelinase-2-deficient mice. J. Lipid Res. 2018, 59, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, M.; Tutusaus, A.; Martinez-Nieto, G.A.; Bárcena, C.; de Gregorio, E.; Moutinho, C.; Barbero-Camps, E.; Villanueva, A.; Colell, A.; Marí, M.; et al. Targeting glucosylceramide synthase upregulation reverts sorafenib resistance in experimental hepatocellular carcinoma. Oncotarget 2016, 7, 8253. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Xu, M.; Liu, P.; Zhang, S.; Shang, R.; Qiao, Y.; Che, L.; Ribback, S.; Cigliano, A.; Evert, K.; et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019, 70, 1600–1613. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.K.K.; Coffelt, S.B.; Cho, C.H.; Wang, X.J.; Lee, C.W.; Chan, F.K.L.; Yu, J.; Sung, J.J.Y. The autophagic paradox in cancer therapy. Oncogene 2012, 31, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Wang, Z.; Jiang, L.; Sui, X.; You, L.; Shou, J.; Jing, Z.; Xie, J.; Ge, W.; Cai, X.; et al. Autophagy inhibition sensitizes hepatocellular carcinoma to the multikinase inhibitor linifanib. Sci. Rep. 2014, 4, 6683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, D.-M.; Wang, G.-L.; Chen, L.; Xu, Y.-Y.; He, S.; Cao, X.-L.; Qin, J.; Zhou, J.-M.; Zhang, Y.-X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.-F.; Li, L.-L.; Lu, J.; Yan, K.; Guo, W.-H.; Zhang, J.-X. XPD Functions as a Tumor Suppressor and Dysregulates Autophagy in Cultured HepG2 Cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 1562–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hou, J.; Wang, X.; Jiang, R.; Yin, Y.; Ji, J.; Deng, L.; Huang, X.; Wang, K.; Sun, B. PTPRO-mediated autophagy prevents hepatosteatosis and tumorigenesis. Oncotarget 2015, 6, 9420–9433. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [Green Version]
- Shu, G.; Xie, B.; Ren, F.; Liu, D.; Zhou, J.; Li, Q.; Chen, J.; Yuan, L.; Zhou, J. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell. Oncol. Dordr. 2013, 36, 121–129. [Google Scholar] [CrossRef]
- Park, M.A.; Zhang, G.; Martin, A.P.; Hamed, H.; Mitchell, C.; Hylemon, P.B.; Graf, M.; Rahmani, M.; Ryan, K.; Liu, X.; et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol. Ther. 2008, 7, 1648–1662. [Google Scholar] [CrossRef] [Green Version]
- Savić, R.; He, X.; Fiel, I.; Schuchman, E.H. Recombinant human acid sphingomyelinase as an adjuvant to sorafenib treatment of experimental liver cancer. PLoS ONE 2013, 8, e65620. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.C.; Walkley, S.U. Niemann-Pick disease, type C and Roscoe Brady. Mol. Genet. Metab. 2017, 120, 34–37. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lafrasse, C.; Rousson, R.; Pentchev, P.G.; Louisot, P.; Vanier, M.T. Free sphingoid bases in tissues from patients with type C Niemann-Pick disease and other lysosomal storage disorders. Biochim. Biophys. Acta 1994, 1226, 138–144. [Google Scholar] [CrossRef]
- Valle, D.; Beaudet, A.L.; Vogelstein, B.; Kinzler, K.W.; Antonarakis, S.E.; Ballabio, A.; Gibson, K.M.; Mitchell, G.A.; FACMG. Lysosomal Disorders. In The Online Metabolic and Molecular Bases of Inherited Disease; David Valle, M., Arthur, L., Beaudet, M., Bert Vogeslstein, M., Kenneth, W., Kinzler, P.D., Stylianos, E., Antonarakis, M.D., Andrea Ballabio, M.K., Michael Gibson, P.D., et al., Eds.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Pick, L., II. Niemann-Pick’s disease and other forms of so-called xanthomatosis. Am. J. Med. Sci. 1933, 185, 615–616. [Google Scholar] [CrossRef]
- Pavlů-Pereira, H.; Asfaw, B.; Poupctová, H.; Ledvinová, J.; Sikora, J.; Vanier, M.T.; Sandhoff, K.; Zeman, J.; Novotná, Z.; Chudoba, D.; et al. Acid sphingomyelinase deficiency. Phenotype variability with prevalence of intermediate phenotype in a series of twenty-five Czech and Slovak patients. A multi-approach study. J. Inherit. Metab. Dis. 2005, 28, 203–227. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of Type A Niemann-Pick disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Desnick, R.J.; Schuchman, E.H.; Hossain, S.; Wallenstein, S.; Lamm, C.; McGovern, M.M. The natural history of type B Niemann-Pick disease: Results from a 10-year longitudinal study. Pediatrics 2004, 114, e672–e677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, M.M.; Wasserstein, M.P.; Giugliani, R.; Bembi, B.; Vanier, M.T.; Mengel, E.; Brodie, S.E.; Mendelson, D.; Skloot, G.; Desnick, R.J.; et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics 2008, 122, e341–e349. [Google Scholar] [CrossRef] [Green Version]
- McGovern, M.M.; Avetisyan, R.; Sanson, B.-J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41. [Google Scholar] [CrossRef]
- Lidove, O.; Belmatoug, N.; Froissart, R.; Lavigne, C.; Durieu, I.; Mazodier, K.; Serratrice, C.; Douillard, C.; Goizet, C.; Cathebras, P.; et al. Déficit en sphingomyélinase acide (maladie de Niemann-Pick B): Une étude rétrospective multicentrique de 28 patients adultes. Rev. Med. Interne 2017, 38, 291–299. [Google Scholar] [CrossRef]
- Cassiman, D.; Packman, S.; Bembi, B.; Turkia, H.B.; Al-Sayed, M.; Schiff, M.; Imrie, J.; Mabe, P.; Takahashi, T.; Mengel, K.E.; et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol. Genet. Metab. 2016, 118, 206–213. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Wasserstein, M.P.; Jones, S.A.; Schiano, T.D.; Cox, G.F.; Puga, A.C. Clearance of Hepatic Sphingomyelin by Olipudase Alfa Is Associated with Improvement in Lipid Profiles in Acid Sphingomyelinase Deficiency. Am. J. Surg. Pathol. 2016, 40, 1232–1242. [Google Scholar] [CrossRef] [Green Version]
- Eskes, E.C.B.; Sjouke, B.; Vaz, F.M.; Goorden, S.M.I.; van Kuilenburg, A.B.P.; Aerts, J.M.F.G.; Hollak, C.E.M. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol. Genet. Metab. 2020, 130, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Jones, S.A.; Soran, H.; Diaz, G.A.; Lippa, N.; Thurberg, B.L.; Culm-Merdek, K.; Shamiyeh, E.; Inguilizian, H.; Cox, G.F.; et al. Successful within-Patient Dose Escalation of Olipudase Alfa in Acid Sphingomyelinase Deficiency. Mol. Genet. Metab. 2015, 116, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Diaz, G.A.; Lachmann, R.H.; Jouvin, M.-H.; Nandy, I.; Ji, A.J.; Puga, A.C. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): Safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 2018, 41, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbins, E.; Palmada, M.; Reichel, M.; Lüth, A.; Böhmer, C.; Amato, D.; Müller, C.P.; Tischbirek, C.H.; Groemer, T.W.; Tabatabai, G.; et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat. Med. 2013, 19, 934–938. [Google Scholar] [CrossRef] [Green Version]
- Hua, G.; Kolesnick, R. Using ASMase Knockout Mice to Model Human Diseases. Sphingolipids Dis. 2012, 216, 29–54. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Canals, D.; Hannun, Y.A. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell. Signal. 2009, 21, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid sphingomyelinase deficient mice: A model of types A and B Niemann-Pick disease. Nat. Genet. 1995, 10, 288–293. [Google Scholar] [CrossRef]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative Stress: A Pathogenic Mechanism for Niemann-Pick Type C Disease. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Sharma, D.K.; Marks, D.L.; Pagano, R.E. Elevated Endosomal Cholesterol Levels in Niemann-Pick Cells Inhibit Rab4 and Perturb Membrane Recycling. Mol. Biol. Cell 2004, 15, 4500–4511. [Google Scholar] [CrossRef]
- Vance, J.E.; Karten, B. Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J. Lipid Res. 2014, 55, 1609–1621. [Google Scholar] [CrossRef] [Green Version]
- Grassme, H.; Jekle, A.; Riehle, A.; Schwarz, H.; Berger, J.; Sandhoff, K.; Kolesnick, R.; Gulbins, E. CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 2001, 276, 20589–20596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charruyer, A.; Grazide, S.; Bezombes, C.; Müller, S.; Laurent, G.; Jaffrézou, J.-P. UV-C light induces raft-associated acid sphingomyelinase and JNK activation and translocation independently on a nuclear signal. J. Biol. Chem. 2005, 280, 19196–19204. [Google Scholar] [CrossRef] [Green Version]
- Baulies, A.; Ribas, V.; Núñez, S.; Torres, S.; Alarcón-Vila, C.; Martínez, L.; Suda, J.; Ybanez, M.D.; Kaplowitz, N.; García-Ruiz, C.; et al. Lysosomal Cholesterol Accumulation Sensitizes to Acetaminophen Hepatotoxicity by Impairing Mitophagy. Sci. Rep. 2016, 5, 18017. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Luberto, C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000, 10, 73–80. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [Green Version]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 2004, 279, 18384–18391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llacuna, L.; Marí, M.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Morales, A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology 2006, 44, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Unal, B.; Ozcan, F.; Tuzcu, H.; Kırac, E.; Elpek, G.O.; Aslan, M. Inhibition of neutral sphingomyelinase decreases elevated levels of nitrative and oxidative stress markers in liver ischemia-reperfusion injury. Redox Rep. Commun. Free Radic. Res. 2017, 22, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Tuzcu, H.; Unal, B.; Kırac, E.; Konuk, E.; Ozcan, F.; Elpek, G.O.; Demir, N.; Aslan, M. Neutral sphingomyelinase inhibition alleviates apoptosis, but not ER stress, in liver ischemia–reperfusion injury. Free Radic. Res. 2017, 51, 253–268. [Google Scholar] [CrossRef]
- Lee, W.M. Acetaminophen and the U.S. Acute Liver Failure Study Group: Lowering the risks of hepatic failure. Hepatology 2004, 40, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Auzinger, G.; Dhawan, A.; Wendon, J. Acute liver failure. Lancet 2010, 376, 190–201. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumaru, K.; Ji, C.; Kaplowitz, N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 2003, 37, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Meyers, L.L.; Beierschmitt, W.P.; Khairallah, E.A.; Cohen, S.D. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 1988, 93, 378–387. [Google Scholar] [CrossRef]
- Jaeschke, H.; Bajt, M.L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 89, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: The protective effect of allopurinol. J. Pharmacol. Exp. Ther. 1990, 255, 935–941. [Google Scholar] [PubMed]
- Kon, K.; Kim, J.-S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Masyuk, A.I.; Masyuk, T.V.; Larusso, N.F. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J. Hepatol. 2013, 59, 621–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, S.; Feng, D.; Zhao, W.-L.; Sui, S.-F. A novel strategy for the invasive toxin: Hijacking exosome-mediated intercellular trafficking. Traffic 2009, 10, 411–424. [Google Scholar] [CrossRef]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.W.; Goldberg, E.M.; Zidovetzki, R. Ceramides perturb the structure of phosphatidylcholine bilayers and modulate the activity of phospholipase A2. Eur. Biophys. J. EBJ 1998, 27, 361–366. [Google Scholar] [CrossRef]
- Gilbert, S.; Loranger, A.; Omary, M.B.; Marceau, N. Keratin impact on PKCδ- and ASMase-mediated regulation of hepatocyte lipid raft size—implication for FasR-associated apoptosis. J. Cell Sci. 2016, 129, 3262–3273. [Google Scholar] [CrossRef] [Green Version]
- Wurdinger, T.; Gatson, N.N.; Balaj, L.; Kaur, B.; Breakefield, X.O.; Pegtel, D.M. Extracellular vesicles and their convergence with viral pathways. Adv. Virol. 2012, 2012, 767694. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Kakizaki, M.; Yamamoto, Y.; Yabuta, S.; Kurosaki, N.; Kagawa, T.; Kotani, A. The immunological function of extracellular vesicles in hepatitis B virus-infected hepatocytes. PLoS ONE 2018, 13, e0205886. [Google Scholar] [CrossRef]
- Sanada, T.; Hirata, Y.; Naito, Y.; Yamamoto, N.; Kikkawa, Y.; Ishida, Y.; Yamasaki, C.; Tateno, C.; Ochiya, T.; Kohara, M. Transmission of HBV DNA Mediated by Ceramide-Triggered Extracellular Vesicles. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.-D. Alkaline sphingomyelinase: An old enzyme with novel implications. Biochim. Biophys. Acta 2006, 1761, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.-D.; Nilsson, A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog. Lipid Res. 2009, 48, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, P.; Xu, S.-C.; Yang, L.; Voss, U.; Ekblad, E.; Wu, Y.; Min, Y.; Hertervig, E.; Nilsson, Å.; et al. Enhanced colonic tumorigenesis in alkaline sphingomyelinase (NPP7) knockout mice. Mol. Cancer Ther. 2015, 14, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wu, J.; Hertervig, E.; Lindgren, S.; Duan, D.; Nilsson, A.; Duan, R.-D. Identification of aberrant forms of alkaline sphingomyelinase (NPP7) associated with human liver tumorigenesis. Br. J. Cancer 2007, 97, 1441–1448. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.-D.; Hindorf, U.; Cheng, Y.; Bergenzaun, P.; Hall, M.; Hertervig, E.; Nilsson, Å. Changes of activity and isoforms of alkaline sphingomyelinase (nucleotide pyrophosphatase phosphodiesterase 7) in bile from patients undergoing endoscopic retrograde cholangiopancreatography. BMC Gastroenterol. 2014, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.D.; Verkade, H.J.; Cheng, Y.; Havinga, R.; Nilsson, A. Effects of bile diversion in rats on intestinal sphingomyelinases and ceramidase. Biochim. Biophys. Acta 2007, 1771, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.D.; Cheng, Y.; Tauschel, H.D.; Nilsson, A. Effects of ursodeoxycholate and other bile salts on levels of rat intestinal alkaline sphingomyelinase: A potential implication in tumorigenesis. Dig. Dis. Sci. 1998, 43, 26–32. [Google Scholar] [CrossRef]
- Cumings, J.N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain J. Neurol. 1948, 71, 410–415. [Google Scholar] [CrossRef]
- Gitlin, J.D. Wilson disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Manzl, C.; Berger, C.; Hofer, B. Oxidative stress, mitochondrial permeability transition, and cell death in Cu-exposed trout hepatocytes. Toxicol. Appl. Pharmacol. 2005, 209, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Ross, S.; O’Brien, P.J. Lysosomal involvement in hepatocyte cytotoxicity induced by Cu(2+) but not Cd(2+). Free Radic. Biol. Med. 2001, 30, 89–97. [Google Scholar] [CrossRef]
- Sokol, R.J.; Twedt, D.; McKim, J.M.; Devereaux, M.W.; Karrer, F.M.; Kam, I.; von Steigman, G.; Narkewicz, M.R.; Bacon, B.R.; Britton, R.S. Oxidant injury to hepatic mitochondria in patients with Wilson’s disease and Bedlington terriers with copper toxicosis. Gastroenterology 1994, 107, 1788–1798. [Google Scholar] [CrossRef]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rübben, H.; et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Protein | Function |
|---|---|---|
| Alcoholic and non-alcoholic steatohepatitis (ASH/NASH) | ASMase | Triggers hepatocellular apoptosis in response to TNF and Fas-induced fulminant liver injury. Is required in alcohol or HFD-induced lipogenesis and macrosteatosis. Is required for ER stress (either alcohol or HFD-induced or autophagy suppression-mediated). Is activated during HSC activation and required for their transdifferentiation to myofibroblast-like cells that promote fibrogenesis. Is a crucial link in the regulation of methionine metabolism and PC homeostasis mediating NASH progression. |
| NSMase | Less characterized in ASH/NASH. Controversial function of TNF-induced hepatocellular apoptosis. | |
| Hepatocellular carcinoma (HCC) | NSMase-1 | Is downregulated in HCC tissues |
| NSMase-2 | Its deficiency promotes liver tumor development by regulating the survival and proliferation of cancer stem-like cells | |
| ASMase | Promotes cell death by increasing ER stress and autophagy | |
| Niemann–Pick A/B (NPA/B) | ASMase | Its deficiency affects lysosomal sphingolipid accumulation, resulting in lipid-loaded foam cells in a wide variety of organs having a severe impact in their correct functioning. Its deficiency impairs cholesterol trafficking causing oxidative stress and affects vesicle trafficking pathways mediated by Rab proteins as well as fusion of the late endosomal/lysosomal compartments Its deficiency alters lysosomal–mitochondrial interactions, involving impaired mitophagy, resulting in mitochondrial dysfunction and overall contributing to disease progression. |
| Ischemia–reperfusion (I/R) liver injury | ASMase | Its inhibition prevents ceramide increase after hepatic I/R injury, attenuating serum ALT levels, hepatocellular necrosis, cytochrome c release and caspase 3 activation. |
| NSMase | Its inhibition decreases enhanced levels of nitrosative and oxidative stress in I/R injury. Its inhibition downregulates apoptotic stimuli during I/R injury | |
| Drug-induced liver injury (DILI) | ASMase | Its deficiency alters lysosomal–mitochondrial interactions, involving impaired mitophagy, resulting in mitochondrial dysfunction and sensitization to APAP hepatotoxicity. |
| Viral hepatitis B (HBV) | ASMase | Is required for the production of HBV-DNA carrying extracellular vesicles (EV), essential for hepatocyte infection. |
| Hepatobiliary diseases | Alk-SMase | Its activity is reduced in the bile and liver from primary sclerosing cholangitis (PSC) patients Bile salt diversion strongly reduces Alk-SMase activity in the small intestinal content, which may also affect intestinal SM digestion |
| Wilson disease | ASMase | Cu2+ triggers hepatocyte apoptosis through activation of ASMase and the release of ceramide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Sphingomyelinases and Liver Diseases. Biomolecules 2020, 10, 1497. https://doi.org/10.3390/biom10111497
Insausti-Urkia N, Solsona-Vilarrasa E, Garcia-Ruiz C, Fernandez-Checa JC. Sphingomyelinases and Liver Diseases. Biomolecules. 2020; 10(11):1497. https://doi.org/10.3390/biom10111497
Chicago/Turabian StyleInsausti-Urkia, Naroa, Estel Solsona-Vilarrasa, Carmen Garcia-Ruiz, and Jose C. Fernandez-Checa. 2020. "Sphingomyelinases and Liver Diseases" Biomolecules 10, no. 11: 1497. https://doi.org/10.3390/biom10111497