Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine



Abstract
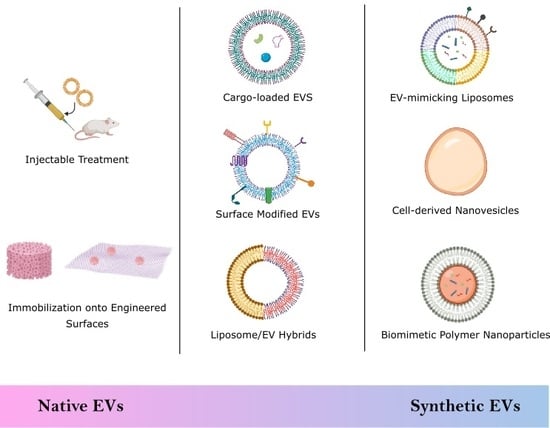
:
1. Introduction
1.1. Exosome Biogenesis
1.2. Exosome Isolation and Characterization
1.3. Physiological Functions of Exosomes and Implications for Regenerative Medicine
2. Biological Stem Cell Derived EVs
2.1. Injectable Treatments
2.2. Engineered Scaffolds and Surfaces
3. Semi-Synthetic EVs
3.1. Drug Delivery
3.1.1. Small Molecules
3.1.2. Nucleic Acids
3.1.3. Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Loading Type | Cargo |
|---|---|---|---|
| Incubation | EVs and cargo are mixed together and coincubated. Cargo molecules follow the concentration gradient to diffuse into EVs. | Passive | Protein [65] Small molecules [60,62] |
| Cell preconditioning or engineering | Cells are cultured in specific environmental conditions, or are transfected/transduced to induce the secretion of cargo-enriched EVs. | Passive | Small molecules [64,67,68] miRNA [70,73,74,75] Protein [79] |
| Freeze/thaw cycle | EV and cargo molecules are incubated together at room temperature and then rapidly frozen at <−80°C and thawed at room temperature. The freezing disrupts the membrane, which allows the cargo molecules to diffuse into the EVs while in a semi-frozen state during the thawing process. | Active | Protein [65] |
| Sonication | A sonicator probe is used to create a mechanical shear force which deforms the EV membrane structure to allow the cargo molecules to diffuse into the EV. | Active | Protein [65] siRNA [82] |
| Extrusion | EVs and cargo molecules are loaded into a syringe and injected forcefully though a porous membrane. The mechanical force disrupts the EV membrane and entraps the cargo within the EV. | Active | Small molecule [83] Protein [65] |
| Chemical permeabilization | Surfactants, or other similar molecules, complex with cholesterol on EV membranes to generate pores, through which cargo molecules can permeate through the EV membrane. | Active | Small molecule [83] Protein [65] |
| Electroporation | EVs are mixed in a solution containing cargo molecules. A voltage is applied that creates temporary pores in the EV membrane which allows these cargo molecules to diffuse into the EVs. When the voltage is removed, the membrane reseals and traps the cargo within the EV. | Active | Small molecule [83,84] siRNA [76] miRNA [69] |
3.2. Surface Modification
3.2.1. Click Chemistry
3.2.2. Integrin-Binding
3.2.3. Phospholipid-Domain Binding
3.3. EV Hybrids
4. Synthetic EVs
4.1. Top-Down Technique
Cell-Derived Nanovesicles
4.2. Bottom-Up Technique
4.2.1. Liposomes
4.2.2. Biomimetic Polymer Nanoparticles
| Synthetic EV Type | Description | Method |
|---|---|---|
| Native EV Modification | Native EVs can be engineered to improve their drug loading, targeting and yield. | Drug loading [60,61,62,63,64,75] |
| Surface peptide modification [91,92,98,99] | ||
| Liposome hybridization [101,102] | ||
| Nanovesicles | Whole cells are fragmented into membrane pieces that self-assemble into vesicles. | Serial extrusion [104,105,106,107] |
| Microfluidics [108] | ||
| Liposomes | Bilayered, spherical lipid vesicles that can be loaded with cargo or modified with surface proteins mimicking native membrane proteins. | Surface-conjugated peptides [113] |
| Tailored lipid formulation [111,112] | ||
| Biomimetic Polymer Nanoparticles | Polymer-based nanoparticles are created and mechanically coated with plasma membrane to create nanovesicles with a core-shell structures. | PLGA core, plasma membrane coating [115,116,120,121] |
| Silicon core, plasma membrane coating [117] |
5. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampogna, G.; Guraya, S.Y.; Forgione, A. Regenerative medicine: Historical roots and potential strategies in modern medicine. J. Microsc. Ultrastruct. 2015, 3, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, G.C.; Callaghan, M.J.; Longaker, M.T. Progress and potential for regenerative medicine. Annu. Rev. Med. 2007, 58, 299–312. [Google Scholar] [CrossRef] [PubMed]
- De Jong, O.G.; Van Balkom, B.W.M.; Schiffelers, R.M.; Bouten, C.V.C.; Verhaar, M.C. Extracellular Vesicles: Potential Roles in Regenerative Medicine. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatima, F.; Nawaz, M. Stem cell-derived exosomes: Roles in stromal remodeling, tumor progression, and cancer immunotherapy. Chin. J. Cancer 2015, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhane, T.N.; Sokic, S.; Schardt, J.S.; Raiker, R.S.; Lin, J.W.; Jay, S.M. Emerging Roles for Extracellular Vesicles in Tissue Engineering and Regenerative Medicine. Tissue Eng. Part B Rev. 2015, 21, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.S.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.G.-E.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, C.; Wei, L.; Li, Q.; Niu, X.; Xu, Y.; Wang, Y.; Zhao, J. Exosomes derived from endothelial progenitor cells attenuate vascular repair and accelerate reendothelialization by enhancing endothelial function. Cytotherapy 2016, 18, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Biancone, L.; Figliolini, F.; Beltramo, S.; Medica, D.; Deregibus, M.C.; Galimi, F.; Romagnoli, R.; Salizzoni, M.; Tetta, C.; et al. Microvesicles derived from endothelial progenitor cells enhance neoangiogenesis of human pancreatic islets. Cell Transpl. 2012, 21, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, M.; Yu, B.; Wang, J.; Wang, Y.; Liu, M.; Paul, C.; Millard, R.W.; Xiao, D.-S.; Ashraf, M.; Xu, M. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget 2017, 8, 45200–45212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Rana, S.; Zöller, M. Host Matrix Modulation by Tumor Exosomes Promotes Motility and Invasiveness. Neoplasia 2013, 15, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albacete-Albacete, L.; Navarro-Lerida, I.; Lopez, J.A.; Martin-Padura, I.; Astudillo, A.M.; Van-Der-Heyden, M.; Balsinde, J.; Orend, G.; Vazquez, J.; del Pozo, M.A. ECM deposition is driven by caveolin1-dependent regulation of exosomal biogenesis and cargo sorting. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nature Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Shenoda, B.B.; Ajit, S.K. Modulation of Immune Responses by Exosomes Derived from Antigen-Presenting Cells. Clin. Med. Insights Pathol. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytom. Part A 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.-Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-‘t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Willis, G.R.; Kourembanas, S.; Mitsialis, S.A. Toward Exosome-Based Therapeutics: Isolation, Heterogeneity, and Fit-for-Purpose Potency. Front. Cardiovasc. Med. 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohde, E.; Pachler, K.; Gimona, M. Manufacturing and characterization of extracellular vesicles from umbilical cord–derived mesenchymal stromal cells for clinical testing. Cytotherapy 2019, 21, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Andriolo, G.; Provasi, E.; Lo Cicero, V.; Brambilla, A.; Soncin, S.; Torre, T.; Milano, G.; Biemmi, V.; Vassalli, G.; Turchetto, L.; et al. Exosomes From Human Cardiac Progenitor Cells for Therapeutic Applications: Development of a GMP-Grade Manufacturing Method. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials – an ISEV position paper. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.J.; Lee, C.; Rojalin, T.; Carney, R.P.; Hazari, S.; Knudson, A.; Lam, K.; Saari, H.; Ibañez, E.L.; Viitala, T.; et al. Single exosome study reveals subpopulations distributed among cell lines with variability related to membrane content. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhu, Y.-L.; Zhou, Y.-Y.; Liang, G.-F.; Wang, Y.-Y.; Hu, F.-H.; Xiao, Z.-D. Exosome Uptake through Clathrin-mediated Endocytosis and Macropinocytosis and Mediating miR-21 Delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Gonda, A.; Kabagwira, J.; Senthil, G.N.; Wall, N.R. Internalization of Exosomes through Receptor-Mediated Endocytosis. Mol. Cancer Res. 2019, 17, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Keshtkar, S.; Azarpira, N.; Ghahremani, M.H. Mesenchymal stem cell-derived extracellular vesicles: Novel frontiers in regenerative medicine. Stem Cell Res. Ther. 2018, 9. [Google Scholar] [CrossRef]
- Zhao, T.; Sun, F.; Liu, J.; Ding, T.; She, J.; Mao, F.; Xu, W.; Qian, H.; Yan, Y. Emerging Role of Mesenchymal Stem Cell-derived Exosomes in Regenerative Medicine. Curr. Stem Cell Res. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Becker, J.C.; Gao, K.; Carney, R.P.; Lankford, L.; Keller, B.A.; Herout, K.; Lam, K.S.; Farmer, D.L.; Wang, A. Neuroprotective effect of placenta-derived mesenchymal stromal cells: Role of exosomes. FASEB J. 2019, 33, 5836–5849. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Zhang, S.; Barthe, S.; Kumar, P.; Pivetti, C.; Kreutzberg, N.; Reed, C.; Wang, Y.; Paxton, Z.; Farmer, D.; et al. Placental Mesenchymal Stem Cell-Derived Extracellular Vesicles Promote Myelin Regeneration in an Animal Model of Multiple Sclerosis. Cells 2019, 8, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Xiao, L.; Peng, J.; Qian, Y.-Q.; Huang, C.-C. Exosomes derived from bone marrow mesenchymal stem cells improve osteoporosis through promoting osteoblast proliferation via MAPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3962–3970. [Google Scholar] [PubMed]
- Aguado, B.A.; Mulyasasmita, W.; Su, J.; Lampe, K.J.; Heilshorn, S.C. Improving viability of stem cells during syringe needle flow through the design of hydrogel cell carriers. Tissue Eng. Part A 2012, 18, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K.L.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6. [Google Scholar] [CrossRef]
- Williams, A.M.; Dennahy, I.S.; Bhatti, U.F.; Halaweish, I.; Xiong, Y.; Chang, P.; Nikolian, V.C.; Chtraklin, K.; Brown, J.; Zhang, Y.; et al. Mesenchymal Stem Cell-Derived Exosomes Provide Neuroprotection and Improve Long-Term Neurologic Outcomes in a Swine Model of Traumatic Brain Injury and Hemorrhagic Shock. J. Neurotrauma 2019, 36, 54–60. [Google Scholar] [CrossRef]
- Webb, R.L.; Kaiser, E.E.; Scoville, S.L.; Thompson, T.A.; Fatima, S.; Pandya, C.; Sriram, K.; Swetenburg, R.L.; Vaibhav, K.; Arbab, A.S.; et al. Human Neural Stem Cell Extracellular Vesicles Improve Tissue and Functional Recovery in the Murine Thromboembolic Stroke Model. Transl. Stroke Res. 2018, 9, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Webb, R.L.; Kaiser, E.E.; Jurgielewicz, B.J.; Spellicy, S.; Scoville, S.L.; Thompson, T.A.; Swetenburg, R.L.; Hess, D.C.; West, F.D.; Stice, S.L. Human Neural Stem Cell Extracellular Vesicles Improve Recovery in a Porcine Model of Ischemic Stroke. Stroke 2018, 49, 1248–1256. [Google Scholar] [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-Mediated Transfer of miR-133b from Multipotent Mesenchymal Stromal Cells to Neural Cells Contributes to Neurite Outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.S.; Xu, Q. Neuronal Differentiation of Human Mesenchymal Stem Cells Using Exosomes Derived from Differentiating Neuronal Cells. PLoS ONE 2015, 10, e0135111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Materials Today 2011, 14, 88–95. [Google Scholar]
- Xie, H.; Wang, Z.; Zhang, L.; Lei, Q.; Zhao, A.; Wang, H.; Li, Q.; Cao, Y.; Jie Zhang, W.; Chen, Z. Extracellular Vesicle-functionalized Decalcified Bone Matrix Scaffolds with Enhanced Pro-angiogenic and Pro-bone Regeneration Activities. Sci. Rep. 2017, 7, 45622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/tricalcium phosphate combination scaffolds can enhance bone regeneration by activating the PI3K/Akt signaling pathway. Stem Cell Res. Ther. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Liu, Y.; Zhang, P.; Tang, Y.; Zhou, M.; Jiang, W.; Zhang, X.; Wu, G.; Zhou, Y. Tissue-Engineered Bone Immobilized with Human Adipose Stem Cells-Derived Exosomes Promotes Bone Regeneration. ACS Appl. Mater. Interfaces 2018, 10, 5240–5254. [Google Scholar] [CrossRef]
- Shi, Q.; Qian, Z.; Liu, D.; Sun, J.; Wang, X.; Liu, H.; Xu, J.; Guo, X. GMSC-Derived Exosomes Combined with a Chitosan/Silk Hydrogel Sponge Accelerates Wound Healing in a Diabetic Rat Skin Defect Model. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Liu, B.; Lee, B.W.; Nakanishi, K.; Villasante, A.; Williamson, R.; Metz, J.; Kim, J.; Kanai, M.; Bi, L.; Brown, K.; et al. Cardiac recovery via extended cell-free delivery of extracellular vesicles secreted by cardiomyocytes derived from induced pluripotent stem cells. Nat. Biomed. Eng. 2018, 2, 293. [Google Scholar] [CrossRef]
- Kang, K.; Ma, R.; Cai, W.; Huang, W.; Paul, C.; Liang, J.; Wang, Y.; Zhao, T.; Kim, H.W.; Xu, M.; et al. Exosomes Secreted from CXCR4 Overexpressing Mesenchymal Stem Cells Promote Cardioprotection via Akt Signaling Pathway following Myocardial Infarction. Stem Cells Int. 2015, 2015, 659890. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as Drug Delivery Vehicles for Parkinson’s Disease Therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Pessina, A.; Bonomi, A.; Coccè, V.; Invernici, G.; Navone, S.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Viganò, L.; Locatelli, A.; et al. Mesenchymal Stromal Cells Primed with Paclitaxel Provide a New Approach for Cancer Therapy. PLoS ONE 2011, 6, e28321. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, A.; Coccè, V.; Cavicchini, L.; Sisto, F.; Dossena, M.; Balzarini, P.; Portolani, N.; Ciusani, E.; Parati, E.; Alessandri, G.; et al. Adipose tissue-derived stromal cells primed in vitro with paclitaxel acquire anti-tumor activity. Int. J. Immunopathol. Pharmacol. 2013, 26, 33–41. [Google Scholar] [CrossRef]
- Pessina, A.; Coccè, V.; Bonomi, A.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Ciusani, E.; Navone, S.; Marfia, G.; Parati, E.; et al. Human skin-derived fibroblasts acquire in vitro anti-tumor potential after priming with Paclitaxel. Anticancer Agents Med. Chem. 2013, 13, 523–530. [Google Scholar]
- Momen-Heravi, F.; Bala, S.; Bukong, T.; Szabo, G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomedicine 2014, 10, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Pan, Y.; Li, X.H.; Yang, X.Y.; Feng, Y.L.; Tan, H.H.; Jiang, L.; Feng, J.; Yu, X.Y. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Disease 2016, 7, e2277. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chen, L.; Guo, X.; Wang, H.; Chen, W.; Wu, G.; Gu, B.; Miao, W.; Kong, J.; Jin, X.; et al. Comparative Analysis of microRNA Expression Profiles of Exosomes Derived from Normal and Hypoxic Preconditioning Human Neural Stem Cells by Next Generation Sequencing. J. Biomed. Nanotechnol. 2018, 14, 1075–1089. [Google Scholar] [CrossRef]
- Gray, W.D.; French, K.M.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of Therapeutic Covariant MicroRNA Clusters in Hypoxia-Treated Cardiac Progenitor Cell Exosomes Using Systems Biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Zhou, J.; Liu, B.; Liang, C.; Pan, X.; Zhang, Y.; Zhang, Y.; Wang, Y.; Shao, L.; Zhu, B.; et al. Delivery of miR-675 by stem cell-derived exosomes encapsulated in silk fibroin hydrogel prevents aging-induced vascular dysfunction in mouse hindlimb. Mater. Sci. Eng. C. Mater. Biol. Appl. 2019, 99, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.-H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Stremersch, S.; Vandenbroucke, R.E.; Van Wonterghem, E.; Hendrix, A.; De Smedt, S.C.; Raemdonck, K. Comparing exosome-like vesicles with liposomes for the functional cellular delivery of small RNAs. J. Control. Release 2016, 232, 51–61. [Google Scholar] [CrossRef]
- O’Loughlin, A.J.; Mäger, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Putz, U.; Howitt, J.; Lackovic, J.; Foot, N.; Kumar, S.; Silke, J.; Tan, S.-S. Nedd4 family-interacting protein 1 (Ndfip1) is required for the exosomal secretion of Nedd4 family proteins. J. Biol. Chem. 2008, 283, 32621–32627. [Google Scholar] [CrossRef] [Green Version]
- Sterzenbach, U.; Putz, U.; Low, L.-H.; Silke, J.; Tan, S.-S.; Howitt, J. Engineered Exosomes as Vehicles for Biologically Active Proteins. Mol. Ther. 2017, 25, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Wu, N.; Yang, J.-M.; Gould, S.J. Protein Targeting to Exosomes/Microvesicles by Plasma Membrane Anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Mokarizadeh, A.; Delirezh, N.; Morshedi, A.; Mosayebi, G.; Farshid, A.-A.; Dalir-Naghadeh, B. Phenotypic modulation of auto-reactive cells by insertion of tolerogenic molecules via MSC-derived exosomes. Vet. Res. Forum 2012, 3, 257–261. [Google Scholar] [PubMed]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased Amyloid-β Pathologies by Intracerebral Loading of Glycosphingolipid-enriched Exosomes in Alzheimer Model Mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presolski, S.I.; Hong, V.P.; Finn, M.G. Copper-Catalyzed Azide–Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol. 2011, 3, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwe, K.; Brechbiel, M.W. Growing Applications of “Click Chemistry” for Bioconjugation in Contemporary Biomedical Research. Cancer Biother. Radiopharm. 2009, 24, 289–302. [Google Scholar] [CrossRef]
- Bonnet, D.; Ilien, B.; Galzi, J.-L.; Riché, S.; Antheaune, C.; Hibert, M. A Rapid and Versatile Method to Label Receptor Ligands Using “Click” Chemistry: Validation with the Muscarinic M1 Antagonist Pirenzepine. Bioconjugate Chem. 2006, 17, 1618–1623. [Google Scholar] [CrossRef]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem. 2014, 25, 1777–1784. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Han, Y.; An, Y.; Ding, Y.; He, C.; Wang, X.; Tang, Q. NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yun, N.; Mun, D.; Kang, J.-Y.; Lee, S.-H.; Park, H.; Park, H.; Joung, B. Cardiac-specific delivery by cardiac tissue-targeting peptide-expressing exosomes. Biochem. Biophys. Res. Commun. 2018, 499, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.E.; Leonard, J.N. Stabilization of Exosome-targeting Peptides via Engineered Glycosylation. J. Biol. Chem. 2015, 290, 8166–8172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, R.P.; Hazari, S.; Rojalin, T.; Knudson, A.; Gao, T.; Tang, Y.; Liu, R.; Viitala, T.; Yliperttula, M.; Lam, K.S. Targeting Tumor-Associated Exosomes with Integrin-Binding Peptides. Adv. Biosyst. 2017, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, D.; Xiao, W.; Liu, R.; Kumar, P.; Li, Y.; Zhou, P.; Guo, F.; Farmer, D.L.; Lam, K.S.; Wang, F.; et al. Discovery and Characterization of a Potent and Specific Peptide Ligand Targeting Endothelial Progenitor Cells and Endothelial Cells for Tissue Regeneration. ACS Chem. Biol. 2017, 12, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Liu, R.; Marik, J.; Wang, X.; Takada, Y.; Lam, K.S. Combinatorial chemistry identifies high-affinity peptidomimetics against α 4 β 1 integrin for in vivo tumor imaging. Nat. Chem. Biol. 2006, 2, 381. [Google Scholar] [CrossRef] [PubMed]
- Kooijmans, S.A.A.; Gitz-Francois, J.J.J.M.; Schiffelers, R.M.; Vader, P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug-and-play approach †Electronic supplementary information (ESI) available. Nanoscale 2018, 10, 2413–2426. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Zhang, T.; He, W.; Jin, H.; Liu, C.; Yang, Z.; Ren, J. Methotrexate-Loaded Extracellular Vesicles Functionalized with Therapeutic and Targeted Peptides for the Treatment of Glioblastoma Multiforme. ACS Appl. Mater. Interfaces 2018, 10, 12341–12350. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Zhang, L.; Ban, L.; Chen, P.; Du, W.; Feng, X.; Liu, B.-F. Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells. ACS Appl. Mater. Interfaces 2017, 9, 27441–27452. [Google Scholar] [CrossRef]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome–Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. (Weinh) 2018, 5. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.-S.; Roh, T.-Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. ACS Nano. 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Kim, S.R.; Kim, D.-K.; Seo, M.W.; Lee, C.; Lee, H.M.; Oh, J.-E.; Choi, E.Y.; Lee, D.-S.; Gho, Y.S.; et al. In Vivo Differentiation of Therapeutic Insulin-Producing Cells from Bone Marrow Cells via Extracellular Vesicle-Mimetic Nanovesicles. ACS Nano. 2015, 9, 11718–11727. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xie, J.; Zhu, J.; Kang, C.; Chiang, C.; Wang, X.; Wang, X.; Kuang, T.; Chen, F.; Chen, Z.; et al. Functional exosome-mimic for delivery of siRNA to cancer: In vitro and in vivo evaluation. J. Control. Release 2016, 243, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, W.; Kim, J.; Yoon, J.; Jeong, D.; Cho, S.; Jeong, H.; Yoon, Y.J.; Kim, S.C.; Gho, Y.S.; Park, J. Large-scale generation of cell-derived nanovesicles. Nanoscale 2014, 6, 12056–12064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.; Jo, W.; Jeong, D.; Kim, J.; Jeong, H.; Park, J. Generation of nanovesicles with sliced cellular membrane fragments for exogenous material delivery. Biomaterials 2015, 59, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Subra, C.; Laulagnier, K.; Perret, B.; Record, M. Exosome lipidomics unravels lipid sorting at the level of multivesicular bodies. Biochimie. 2007, 89, 205–212. [Google Scholar] [CrossRef]
- Lu, M.; Zhao, X.; Xing, H.; Xun, Z.; Zhu, S.; Lang, L.; Yang, T.; Cai, C.; Wang, D.; Ding, P. Comparison of exosome-mimicking liposomes with conventional liposomes for intracellular delivery of siRNA. Int. J. Pharm. 2018, 550, 100–113. [Google Scholar] [CrossRef]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11, 20140459. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.-L.; Wang, Y.-J.; Sun, J.; Zhou, J.; Xing, C.; Huang, G.; Li, J.; Yang, H. Artificial chimeric exosomes for anti-phagocytosis and targeted cancer therapy. Chem. Sci. 2019, 10, 1555–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.-M.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Ding, Y.; Zhang, Y.; Wang, B.; Zhao, X.; Cheng, K.; Huang, Y.; Taleb, M.; Zhao, J.; Dong, W.-F.; et al. Surface Functionalization of Polymeric Nanoparticles with Umbilical Cord-Derived Mesenchymal Stem Cell Membrane for Tumor-Targeted Therapy. ACS Appl. Mater. Interfaces 2018, 10, 22963–22973. [Google Scholar] [CrossRef]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Tang, J.; Nishi, K.; Yan, C.; Dinh, P.-U.; Cores, J.; Kudo, T.; Zhang, J.; Li, T.-S.; Cheng, K. Fabrication of Synthetic Mesenchymal Stem Cells for the Treatment of Acute Myocardial Infarction in Mice. Circ. Res. 2017, 120, 1768–1775. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Shen, D.; Caranasos, T.G.; Wang, Z.; Vandergriff, A.C.; Allen, T.A.; Hensley, M.T.; Dinh, P.-U.; Cores, J.; Li, T.-S.; et al. Therapeutic microparticles functionalized with biomimetic cardiac stem cell membranes and secretome. Nat. Commun. 2017, 8, 13724. [Google Scholar] [CrossRef]
- Zhang, Q.; Dehaini, D.; Zhang, Y.; Zhou, J.; Chen, X.; Zhang, L.; Fang, R.H.; Gao, W.; Zhang, L. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotech. 2018, 13, 1182–1190. [Google Scholar] [CrossRef]
- Jin, J.; Krishnamachary, B.; Barnett, J.D.; Chatterjee, S.; Chang, D.; Mironchik, Y.; Wildes, F.; Jaffee, E.M.; Nimmagadda, S.; Bhujwalla, Z.M. Human Cancer Cell Membrane-Coated Biomimetic Nanoparticles Reduce Fibroblast-Mediated Invasion and Metastasis and Induce T-Cells. ACS Appl. Mater. Interfaces 2019, 11, 7850–7861. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramasubramanian, L.; Kumar, P.; Wang, A. Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules 2020, 10, 48. https://doi.org/10.3390/biom10010048
Ramasubramanian L, Kumar P, Wang A. Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules. 2020; 10(1):48. https://doi.org/10.3390/biom10010048
Chicago/Turabian StyleRamasubramanian, Lalithasri, Priyadarsini Kumar, and Aijun Wang. 2020. "Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine" Biomolecules 10, no. 1: 48. https://doi.org/10.3390/biom10010048