
Obesity and Its Multiple Clinical Implications between Inflammatory States and Gut Microbiotic Alterations

Abstract
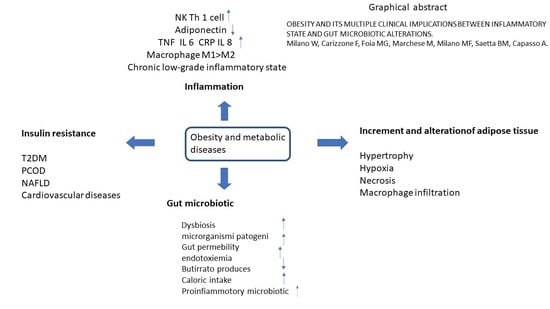
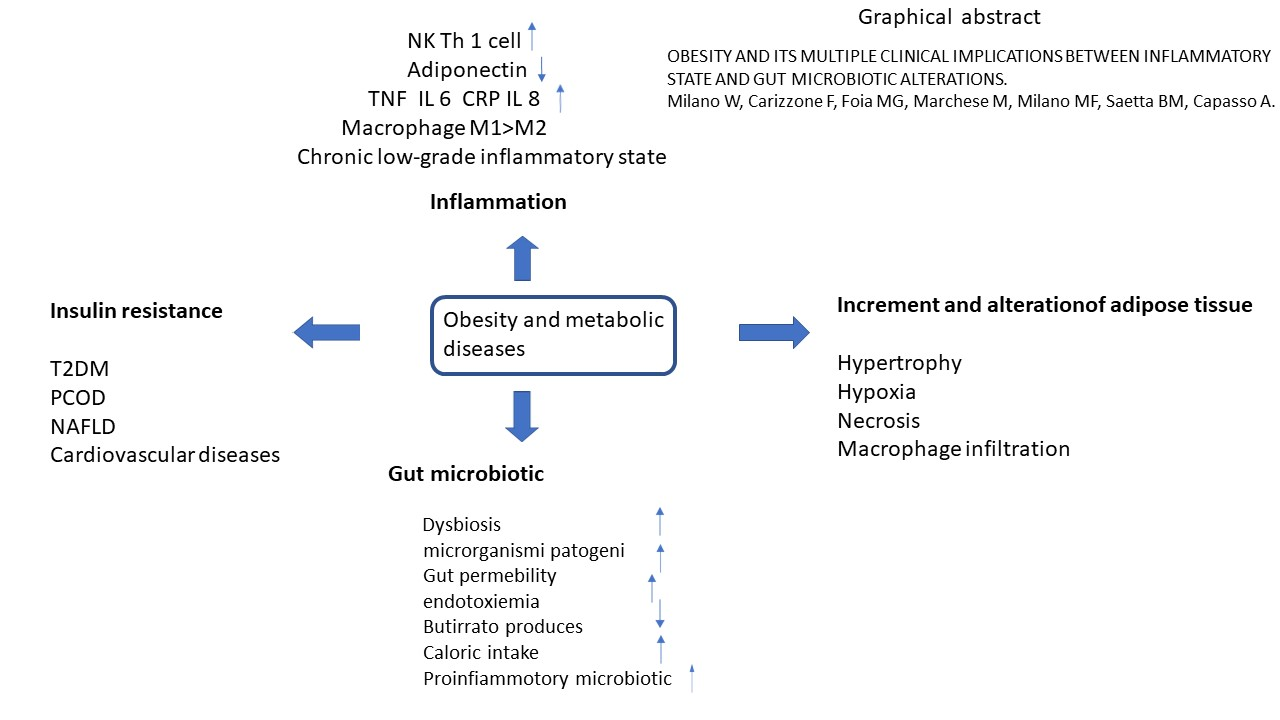
:
{kind=link}
1. Introduction
2. Inflammation
2.1. Activation of the Innate Immune System in Obesity
2.2. Microbiota
3. Discussion
3.1. Therapeutic Potential of Remodeling the Microbiotic Profile
3.2. Remodeling of the Microbiota Secondary to Bariatric Surgery
3.3. Remodulation of the Microbiota-Fecal Microbiota Transplant (FMT) and Gut Microbiota Transplant (GMT)
3.4. Remodeling of the Microbiota through Prebiotics and Probiotics
3.5. Remodeling of the Microbiota through Physical Exercise
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Milano:, W.; Carizzone, F.; De Biasio, V.; Mercorio, M.A.; Milano, M.F.; Saetta, B.; Capasso, A. Neurobiological correlates shared between obesity, bed and food addiction. Endocr. Metab. Immune Disord. Drug Targets 2022, 360, 1–8. [Google Scholar]
- Izaola, O.; de Luis, D.; Sajoux, I.; Domingo, J.C.; Vidal, M. Inflamación y obesidad [lipoinflamación]. Nutr. Hosp. 2015, 31, 2352–2358. [Google Scholar] [PubMed] [Green Version]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Role of Gut Microbiota in the Aetiology of Obesity: Proposed Mechanisms and Review of the Literature. J. Obes. 2016, 2016, 7353642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.B.; Smith, M.S. Obesity statistics. Prim. Care 2016, 43, 121–135. [Google Scholar] [CrossRef]
- Popkin, B.M.; Adair, L.S.; Ng, S.W. Global nutrition transition and the pandemic of obesity in developing countries. Nutr. Rev. 2012, 70, 3–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyts, P.; Delzenne, N. Editorial: The Brain—Gut—Microbiome Network in Metabolic Regulation and Dysregulation. Front. Endocrinol. 2021, 12, 760558. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Overweight and Obesity: Adult Obesity Facts. Available online: https://www.cdc.gov/obesity/data/adult.html (accessed on 28 August 2021).
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity among Adults; NCHS Data Brief; US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics: Washington, DC, USA, 2020; Volume 360, pp. 1–8.
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Nguyen, D.M.; El-Serag, H.B. The epidemiology of obesity. Gastroenterol. Clin. North Am. 2010, 39, 1–7. [Google Scholar] [CrossRef]
- World Health Organization. Obesity. 2015. Available online: http://www.who.int/topics/obesity/en/ (accessed on 26 June 2015).
- Kobyliak, N.; Conte, C.; Cammarota, G.; Haley, A.P.; Styriak, I.; Gaspar, L.; Fusek, J.; Rodrigo, L.; Kruzliak, P. Probiotics in prevention and treatment of obesity: A critical view. Nutr. Metab. 2016, 13, 14. [Google Scholar] [CrossRef] [Green Version]
- American Medical Association House of Delegates. Resolution 420 [A-13]. 2013. Available online: http://www.npr.org/documents/2013/jun/amaresolution-obesity.pdf (accessed on 26 June 2015).
- Pataky, Z.; Bobbioni-Harsch, E.; Golay, A. Open questions about metabolically normal obesity. Int. J. Obes. 2010, 34 (Suppl. S2), S18–S23. [Google Scholar] [CrossRef] [Green Version]
- Barazzoni, R.; Cappellari, G.G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight. Disord. Stud. Anorex. Bulim. Obes. 2018, 23, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Leisegang, K.; Sengupta, P.; Agarwal, A.; Henkel, R. Obesity and male infertility: Mechanisms and management. Androgenic 2021, 53, e13617. [Google Scholar] [CrossRef]
- Wofford, M.R.; King, D.S.; Harrell, T.K. Drug-induced metabolic syndrome. J. Clin. Hypertens. 2006, 8, 114–119. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [Green Version]
- Tchernof, A.; Despres, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef] [Green Version]
- Spencer, M.; Yao-Borengasser, A.; Unal, R.; Rasouli, N.; Gurley, C.M.; Zhu, B.; Peterson, C.A.; Kern, P.A. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am. J. Physiol. Metab. 2010, 299, E1016–E1027. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Samaras, K.; Botelho, N.K.; Chisholm, D.J.; Lord, R.V. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity 2010, 18, 884–889. [Google Scholar] [CrossRef]
- Chatterjee, T.K. Proinflammatory phenotype of perivascular adipocytes: Influence of high-fat feeding. Circ. Res. 2009, 104, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced tyrosine kinase activity of the insulin receptor in obesitydiabetes: Central role of tumor necrosis factor-alpha. J. Clin. Investig. 1994, 94, 1543–1549. [Google Scholar] [CrossRef]
- Kern, P.A.; Di Gregorio, G.B.; Lu, T.; Rassouli, N.; Ranganathan, G. Adiponectin expression from human adipose tissue: Relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003, 52, 1779–1785. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Pontillo, A.; Ciotola, M. Weight loss reduces interleukin-18 levels in obese women. J. Clin. Endocrinol. Metab. 2002, 87, 3864–3866. [Google Scholar] [CrossRef]
- Wang, X.; Bao, W.; Liu, J. Infammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Giugliano, D. The metabolic syndrome and infammation: Association or causation? Nutr. Metab. Cardiovasc. Dis. 2004, 14, 228–232. [Google Scholar] [CrossRef]
- Maiorino, M.I.; Bellastella, G.; Giugliano, D. Esposito from infammation to sexual dysfunctions: A journey through diabetes, obesity, and metabolic syndrome. J. Endocrinol. Investig. 2018, 41, 1249–1258. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Invest. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest. 2017, 127, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 14, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Hilde, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 2546. [Google Scholar] [CrossRef]
- Emilsson, V. Genetics of gene expression and its effect on disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef]
- Odegaard, J.I. Alternative M2 activation of Kupffer cells by PPAR delta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Del Proposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatio temporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J. Clin. Endocrinol. Metab. 2009, 94, 4619–4623. [Google Scholar] [CrossRef] [PubMed]
- Plomgaard, P.; Nielsen, A.R.; Fischer, C.P.; Mortensen, O.H.; Broholm, C.; Penkowa, M. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia 2007, 50, 2562–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nagai, Y.; Takatsu, K. Activation and regulation of the pattern recognition receptors in obesity-induced adipose tissue inflammation and insulin resistance. Nutrients 2013, 5, 3757–3778. [Google Scholar] [CrossRef] [Green Version]
- Boulenouar, S.; Michelet, X.; Duquette, D.; Alvarez, D.; Hogan, A.E.; Dold, C. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity 2017, 46, 273–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, T.E.; Rapp, M.; Fan, X.; Weizman, O.-E.; Bhardwaj, P.; Adams, N.M. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity 2016, 45, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Kang, Y.E.; Kim, J.M.; Joung, K.H.; Lee, J.H.; You, B.R.; Choi, M.J. The roles of adipokines, proinflammatory cytokines, and adipose tissue macrophages in obesity-associated insulin resistance in modest obesity and early metabolic dysfunction. PLoS ONE 2016, 11, e0154003. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Pimentel, G.D.; Ganeshan, K.; Carvalheira, J.B. Hypothalamic inflammation and the central nervous system control of energy homeostasis. Mol. Cell. Endocrinol. 2014, 397, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgeryinduced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [Green Version]
- Selvin, E.; Paynter, N.P.; Erlinger, T.P. The effect of weight loss on C-reactive protein: A systematic review. Arch. Intern. Med. 2007, 167, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, C.S.; Redman, L.M. Adipose tissue inflammation and metabolic dysfunction: A clinical perspective. Horm. Mol. Biol. Clin. Invest. 2013, 15, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef]
- Brooks, G.C.; Blaha, M.J.; Blumenthal, R.S. Relation of C-reactive protein to abdominal adiposity. Am. J. Cardiol. 2010, 106, 56–61. [Google Scholar] [CrossRef]
- Leisegang, K.; Henkel, R.; Agarwal, A. Obesity and metabolic syndrome associated with systemic inflammation and the impact on the male reproductive system. Am. J. Reprod. Immunol. 2019, 82, e13178. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; DeBoy, R.T. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; De Angelis, M.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.-H.; Di Ciaula, A. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines 2022, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gutmicrobiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wei, C. Diet, Gut Microbiota and Obesity. J. Nutrition. Health Food Sci. 2015, 3, 1–6. [Google Scholar] [CrossRef]
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [Green Version]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Scarpellini, E.; Capasso, R. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef] [Green Version]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133 (Suppl. S7), 2485S–2493S. [Google Scholar] [CrossRef] [Green Version]
- Caricilli, A.M.; Picardi, P.K.; de Abreu, L.L.; Ueno, M.; Prada, P.O.; Ropelle, E.R.; Hirabara, S.M.; Castoldi, A.; Vieira, P.; Camara, N.O.; et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011, 9, e1001212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, J.A.; Reimer, R.A. Prebiotic fiber modulation of the gut microbiota improves risk factors for obesity and the metabolic syndrome. Gut Microbes 2012, 3, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Fermentation in the human large intestine: Its physiologic consequences and the potential contribution of prebiotics. J. Clin. Gastroenterol. 2011, 45, S120–S127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Lahham, S.H.; Peppelenbosch, M.P.; Roelofsen, H.; Vonk, R.J.; Venema, K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim. Biophys. Acta 2010, 1801, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 81461, 1282–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Antiinflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Khan, S.; Luck, H.; Winer, S.; Winer, D.A. Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat. Commun. 2021, 12, 2598. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, X.; Zhao, Y. Interleukin-33 Promotes REG3γ Expression in Intestinal Epithelial Cells and Regulates Gut Microbiota. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ota, N.; Manzanillo, P.; Kates, L.; Zavala-Solorio, J.; Eidenschenk, C.; Zhang, J.; Lesch, J.; Lee, W.P.; Ross, J. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014, 514, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Conterno, L.; Fava, F.; Viola, R.; Tuohy, K.M. Obesity and the gut microbiota: Does up-regulating colonic fermentation protect against obesity and metabolic disease? Genes Nutr. 2011, 6, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Selber-Hnatiw, S.; Sultana, T.; Tse, W.; Abdollahi, N. Metabolic networks of the human gut microbiota. Microbiology 2020, 166, 96–119. [Google Scholar] [CrossRef]
- Walker, A.W.; Ince, J.; Duncan, S.H. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Thijs, C.; Vink, C. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Gut Microbiota: The Neglected Endocrine Organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassi, M.; Ashafian, F.; Khezerloo, J.K. Changes in gut microbiota and hormones after bariatric surgery: A bench to bedside review. Obes. Surg. 2019, 29, 1663–1674. [Google Scholar] [CrossRef]
- Peck, B.C.E.; Seeley, R.J. How does ‘metabolic surgery’ work its magic? New evidence for gut microbiota. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 81–86. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, Z.-P.; Liu, C.-Q.; Qi, L.; Sheng, Y. Modulation of the gut microbiome: A systematic review of the effect of bariatric surgery. Eur. J. Endocrinol. 2018, 178, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Werling, M.; Fändriks, L.; Björklund, P.; Maleckas, A.; Brandberg, J. Long-term results of a randomized clinical trial comparing Roux-en-Y gastric bypass with vertical banded gastroplasty. Br. J. Surg. 2013, 100, 222–230. [Google Scholar] [CrossRef]
- Tremaroli, V.; Karlsson, F.; Werling, M.; Ståhlman, M.; Kovatcheva-Datchary, P. Roux-En-Y gastric bypass and vertical banded gastroplasty induce long-term changes on the human gut microbiome contributing to fat mass regulation. Cell Metab. 2015, 22, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furet, J.P.; Kong, L.C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef]
- Kelly, C.R.; Khoruts, A.; Staley, C.; Sadowsky, M.J.; Abd, M. Effect of Fecal Microbiota Transplantation on Recurrence in Multiply Recurrent Clostridium difficile Infection. Ann. Intern. Med. 2016, 165, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, M.; Aroniadis, O.C.; Brandt, L.J.; Kelly, C.; Freeman, S. The long-term efficacy and safety of fecal microbiota transplant for recurrent, severe, and complicated Clostridium difficile infection in 146 elderly individuals. J. Clin. Gastroenterol. 2016, 50, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Rubin, D.T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohn’s Colitis 2014, 8, 1569–1581. [Google Scholar] [CrossRef] [Green Version]
- Jayasinghe, T.N.; Chiavaroli, V.; Holland, D.J.; Cutfield, W.S.; O’Sullivan, J.M. The new era of treatment for obesity and metabolic disorders: Evidence and expectations for gut microbiome transplantation. Front. Cell. Infect. Microbiol. 2016, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef]
- Bidu, C.; Escoula, Q.; Bellenger, S.; Spor, A.; Galan, M. The transplantation of ω3 PUFA–Altered gut microbiota of fat-1 mice to wild-type littermates prevents obesity and associated metabolic disorders. Diabetes 2018, 67, 1512–1523. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Ma, C.; Han, L.; Nawaz, M.; Gao, F. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr. Microbiol. 2010, 61, 69–78. [Google Scholar] [CrossRef]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V. Roseburia spp.: A marker of health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef]
- Barczynska, R.; Bandurska, K.; Slizewska, K.; Litwin, M.; Szalecki, M. Intestinal microbiota, obesity and prebiotics. Pol. J. Microbiol. 2015, 64, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Delzenne, N.M.; Kok, N.N. Biochemical basis of oligofructose-induced hypolipidemia in animal models. J. Nutr. 1999, 129, 1467S–1470S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M. Selective increases of bifidobacteria in gut microflora improve highfat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delzenne, N.M.; Kok, N. Effects of fructans-type prebiotics on lipid metabolism. Am. J. Clin. Nutr. 2001, 73 (Suppl. S2), 456S–458S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delzenne, N.M.; Kok, N. Effect of non-digestible fermentable carbohydrates on hepatic fatty acid metabolism. Biochem. Soc. Trans. 1998, 26, 228–230. [Google Scholar] [CrossRef] [Green Version]
- Tazoe, H.; Otomo, Y.; Karaki, S.; Kato, I.; Fukami, Y.; Terasaki, M. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed. Res. 2009, 30, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Karaki, S.; Tazoe, H.; Hayashi, H.; Kashiwabara, H.; Tooyama, K.; Suzuki, Y. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J. Mol. Histol. 2008, 39, 135–142. [Google Scholar] [CrossRef]
- Zhou, J.; Martin, R.J.; Tulley, R.T.; Raggio, A.M.; McCutcheon, K.L.; Shen, L. Dietary resistant starch upregulates total GLP-1 and PYY in a sustained day-long manner through fermentation in rodents. Am. J. Physiol. Endocrinol. Metab. 2008, 295, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Inoue, R.; Tsukahara, T.; Ushida, K.; Chiji, H. Voluntary running exercise alters microbiota composition and increases n-butyrate concentration in the rat cecum. Biosci. Biotechnol. Biochem. 2008, 72, 572–576. [Google Scholar] [CrossRef]
- Evans, C.C.; LePard, K.J.; Kwak, J.W.; Stancukas, M.C.; Laskowski, S. Exercise prevents weight gain and alters the gut microbiota in a mouse model of high fat diet-induced obesity. PLoS ONE 2014, 9, e92193. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Myslicki, J.P.; Bomhof, M.R.; Belke, D.D.; Shearer, J. Exercise training modifies gut microbiota in normal and diabetic mice. Appl. Physiol. Nutr. Metab. 2015, 40, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Denou, E.; Marcinko, K.; Surette, M.G.; Steinberg, G.R.; Schertzer, J.D. High-Intensity exercise training increases the diversity and metabolic capacity of the mouse distal gut microbiota during diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E982–E993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.C.; Wisniewski, P.J.; Noji, M.; McGuinness, L.R.; Häggblom, M.M. The effect of diet and exercise on intestinal integrity and microbial diversity in mice. PLoS ONE 2016, 11, e0150502. [Google Scholar] [CrossRef] [Green Version]
- Murtaza, N.; Burke, L.; Vlahovich, N.; Charlesson, B.; O’Neill, H. Effects of dietary pattern during intensified training on stool microbiota of elite race walkers. Nutrients 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Bressa, C.; Bailén-Andrino, M.; Pérez-Santiago, J.; GonzálezSoltero, R.; Pérez, M. Differences in gut microbiota profile between women with active lifestyle and sedentary women. PLoS ONE 2017, 12, e0171352. [Google Scholar] [CrossRef] [Green Version]
- Sanmiguel, C.; Gupta, A.; Mayer, E.A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Curr. Obes. Rep. 2015, 4, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Muscogiuri, G.; Balercia, G.; Barrea, L.; Cignarelli, A.; Giorgino, F.; Holst, J.J.; Laudisio, D.; Orio, F.; Tirabassi, G.; Colao, A. Gut: A key player in the pathogenesis of type 2 diabetes? Crit. Rev. Food Sci. Nutr. 2018, 58, 1294–1309. [Google Scholar] [CrossRef]
- Alex, S.; Lichtenstein, L.; Dijk, W.; Mensink, R.P.; Tan, N.S.; Kersten, S. ANGPTL4 is produced by entero-endocrine cells in the human intestinal tract. Histochem. Cell Biol. 2014, 141, 383–391. [Google Scholar]
- Yadav, A.N.; Verma, P.; Kumar, R.; Kumar, S.; Kumar, V.; Kumar, K. Probiotic microbes: Biodiversity, mechanisms of action and potential role in human health. In Proceedings of the National Conference on Advances in Food Science and Technology, Paris, France, 23–25 October 2017; p. 33. [Google Scholar]
- Rasmussen, T.S.; Mentzel, C.M.J.; Kot, W.; Castro-Mejia, J.L.; Zuffa, S.; Swann, J.R. Faecal virome transplantation decreases symptoms of type 2 diabetes and obesity in a murine model. Gut 2020, 69, 1–9. [Google Scholar] [CrossRef]
- Huseyin, C.E.; O’Toole, P.W.; Cotter, P.D.; Scanlan, P.D. Forgotten fungi—Thegut mycobiome in human health and disease. FEMS Microbiol. Rev. 2017, 41, 479–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appari, M.; Channon, K.M.; McNeill, E. Channon and Eileen McNeill Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Rebello, O.D.; Henne, A.; Nikolka, F.; Klein, T.; Maedler, K. GLP-2 Is Locally Produced From Human Islets and Balances Inflammation Through an Inter-Islet-Immune Cell Crosstalk. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milano, W.; Carizzone, F.; Foia, M.; Marchese, M.; Milano, M.; Saetta, B.; Capasso, A. Obesity and Its Multiple Clinical Implications between Inflammatory States and Gut Microbiotic Alterations. Diseases 2023, 11, 7. https://doi.org/10.3390/diseases11010007
Milano W, Carizzone F, Foia M, Marchese M, Milano M, Saetta B, Capasso A. Obesity and Its Multiple Clinical Implications between Inflammatory States and Gut Microbiotic Alterations. Diseases. 2023; 11(1):7. https://doi.org/10.3390/diseases11010007
Chicago/Turabian StyleMilano, Walter, Francesca Carizzone, Mariagabriella Foia, Magda Marchese, Mariafrancesca Milano, Biancamaria Saetta, and Anna Capasso. 2023. "Obesity and Its Multiple Clinical Implications between Inflammatory States and Gut Microbiotic Alterations" Diseases 11, no. 1: 7. https://doi.org/10.3390/diseases11010007