
Theoretical DFT Investigation of Structure and Electronic Properties of η5-Cyclopentadienyl Half-Sandwich Organochalcogenide Complexes


Abstract
:1. Introduction
2. Computational Methods
3. Result and Discussion
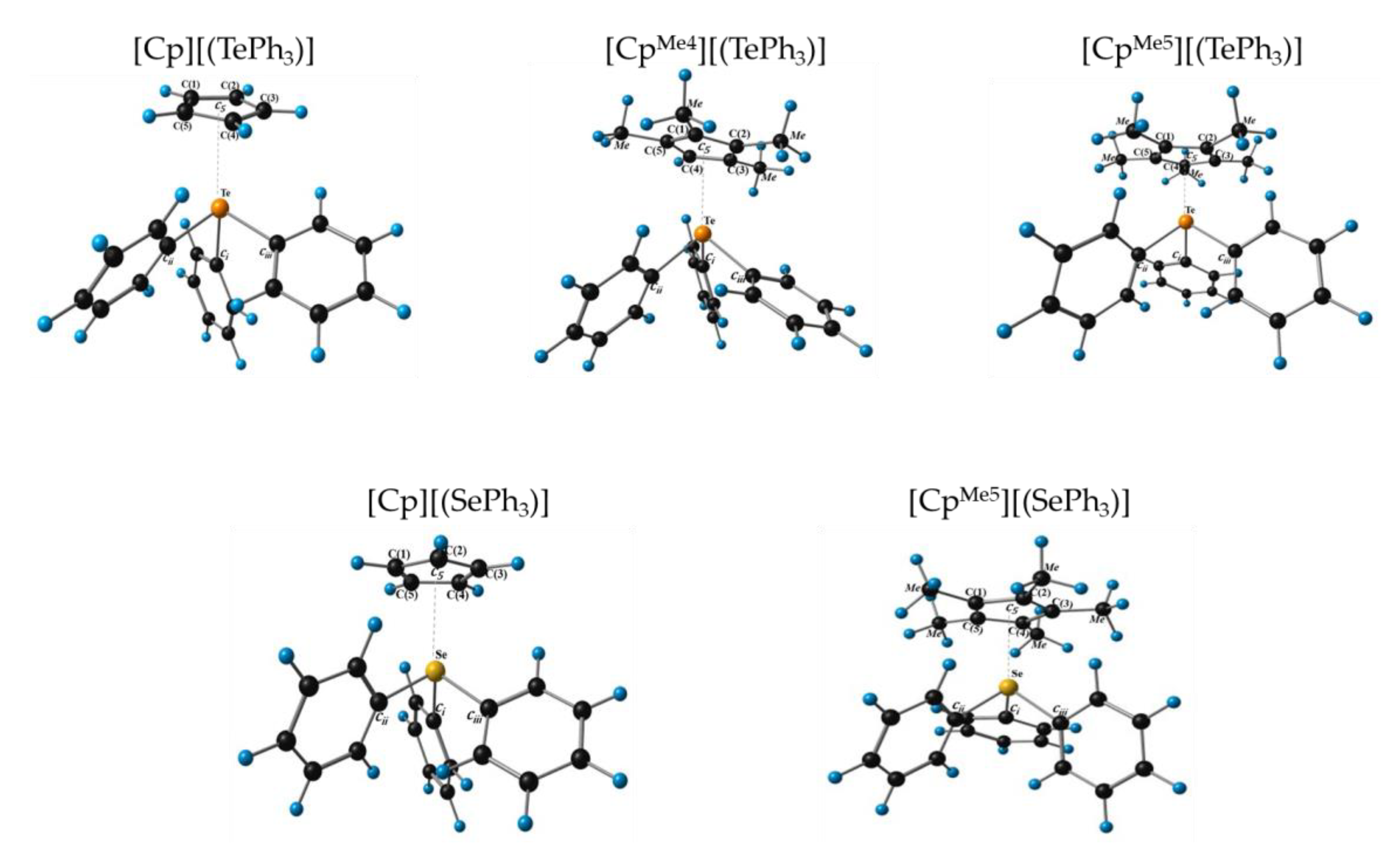
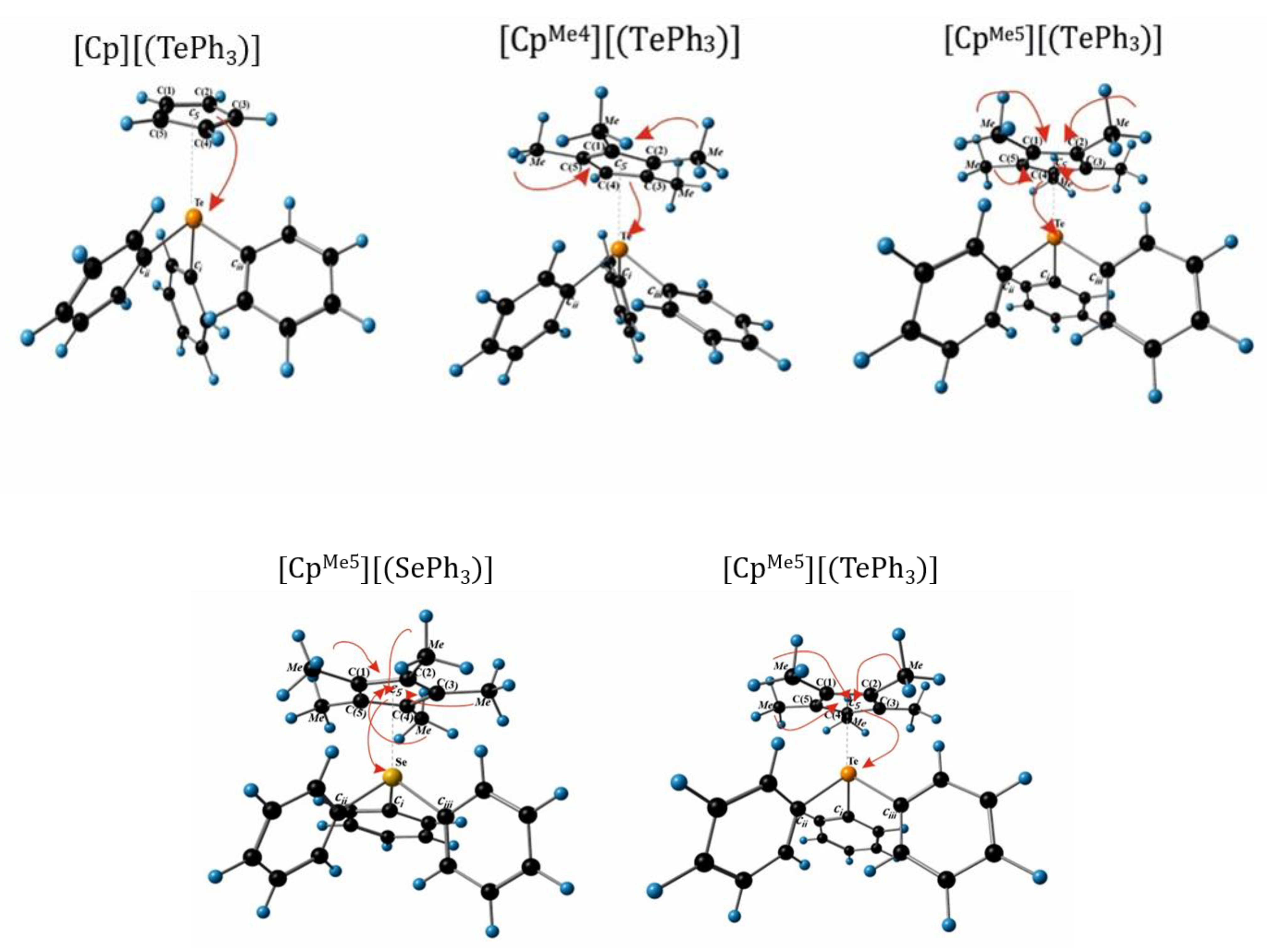
3.1. Structure and Stability η5-Cyclopentadienyl Chalcogenide Complexes
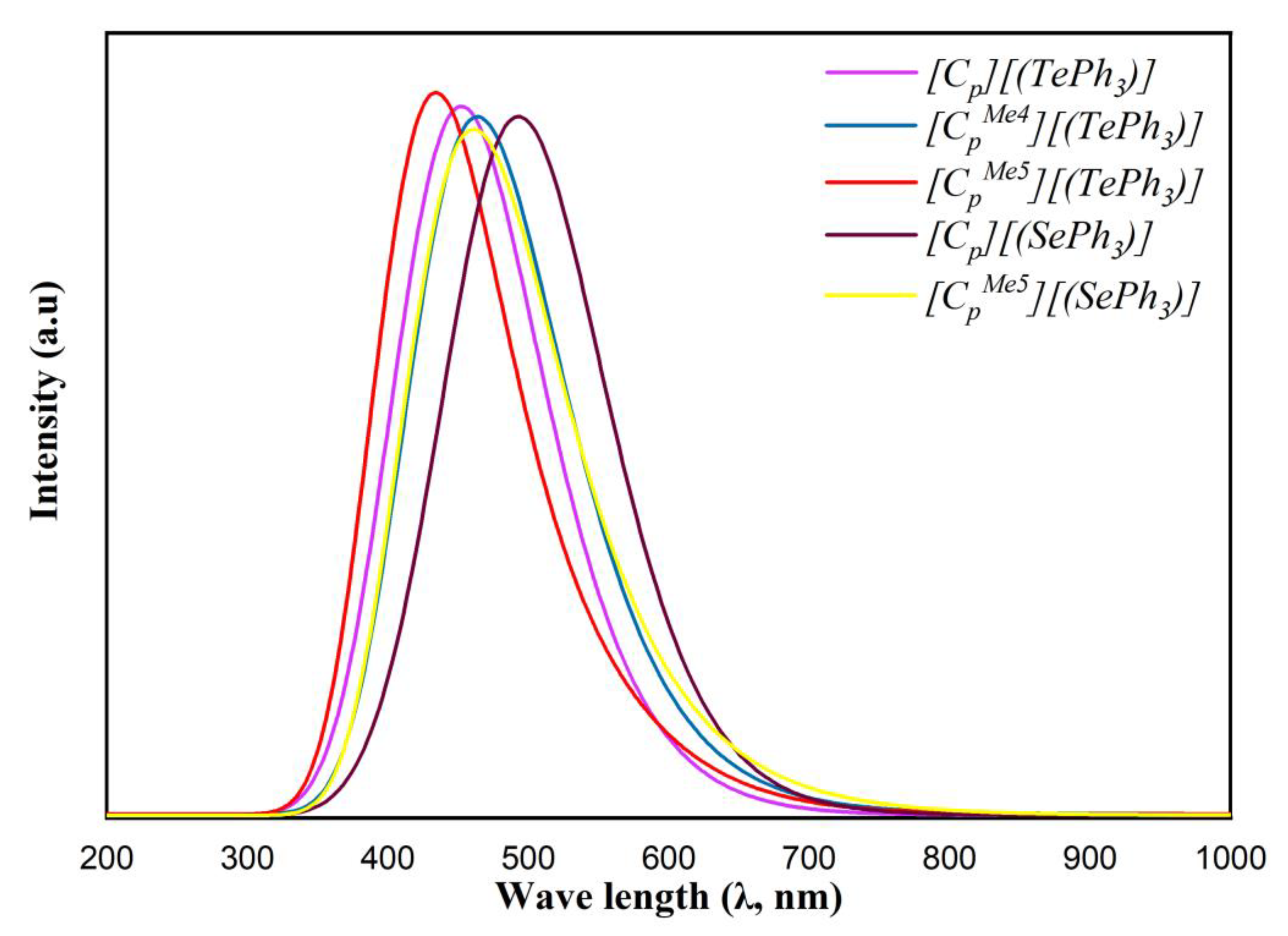
3.2. Electronic Structure of η5-Cyclopentadienyl Organochalcogenide Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, S.-S.; Ziller, J.W.; Zhang, Y.-Q.; Wang, B.-W.; Evans, W.J.; Gao, S. A half-sandwich organometallic single-ion magnet with hexamethylbenzene coordinated to the Dy (III) ion. Chem. Commun. 2014, 50, 11418–11420. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, H.-N.; Jin, G.-X. Molecular borromean rings based on half-sandwich organometallic rectangles. Acc. Chem. Res. 2018, 51, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, J.P.; Pape, V.F.; Szakács, G.; Németi, G.; Dénes, M.; Holczbauer, T.; May, N.V.; Enyedy, É.A. Half-sandwich organometallic Ru and Rh complexes of (N, N) donor compounds: Effect of ligand methylation on solution speciation and anticancer activity. Dalton Trans. 2021, 50, 8218–8231. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, M.; Li, X.; Zhong, W.; Wang, W.; Xiao, Z.; Jiang, X.; Lu, C.; Liu, X. Three half-sandwiched iron (II) monocarbonyl complexes with PNP ligands: Their chemistry upon reduction and catalysis on proton reduction. Electrochim. Acta 2022, 433, 141207. [Google Scholar] [CrossRef]
- Kisets, I.; Zabelinskaya, S.; Gelman, D. Synthesis and Catalytic Properties of a Carbometalated Half-Sandwich Ru (II) Complex Bearing a Rigid Polyaromatic Tether. Organometallics 2021, 41, 76–82. [Google Scholar] [CrossRef]
- Pilia, L.; Shuku, Y.; Dalgleish, S.; Hofmann, D.W.; Melis, N.; Awaga, K.; Robertson, N. Effect of fluorination on the crystal and electronic structure of organometallic cyclopentadienyl-phenylenediamino-cobalt complexes. J. Organomet. Chem. 2020, 918, 121277. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, W.; Guan, A.-L.; Liu, S.; Yao, Z.-J. Half-Sandwich Iridium Complexes Based on β-Ketoamino Ligands: Preparation, Structure, and Catalytic Activity in Amide Synthesis. Inorg. Chem. 2021, 60, 11514–11520. [Google Scholar] [CrossRef]
- Avramov, P.; Sakai, S.; Naramoto, H.; Narumi, K.; Matsumoto, Y.; Maeda, Y. Theoretical DFT Study of Atomic Structure and Spin States of the Co x (C60) n (x= 3− 8, n= 1, 2) Complex Nanoclusters. J. Phys. Chem. C 2008, 112, 13932–13936. [Google Scholar] [CrossRef]
- Avramov, P.; Naramoto, H.; Sakai, S.; Narumi, K.; Lavrentiev, V.; Maeda, Y. Quantum Chemical Study of Atomic Structure Evolution of Co x/C60 (x≤ 2.8) Composites. J. Phys. Chem. A 2007, 111, 2299–2306. [Google Scholar] [CrossRef]
- Sakai, S.; Naramoto, H.; Avramov, P.V.; Yaita, T.; Lavrentiev, V.; Narumi, K.; Baba, Y.; Maeda, Y. Comparative study of structures and electrical properties in cobalt–fullerene mixtures by systematic change of cobalt content. Thin Solid Film. 2007, 515, 7758–7764. [Google Scholar] [CrossRef]
- Sakai, S.; Yakushiji, K.; Mitani, S.; Sugai, I.; Takanashi, K.; Naramoto, H.; Avramov, P.V.; Lavrentiev, V.; Narumi, K.; Maeda, Y. Magnetic and magnetotransport properties in nanogranular Co/C60-Co film with high magnetoresistance. Mater. Trans. 2007, 48, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Sadler, P.J. Transfer hydrogenation catalysis in cells. RSC Chem. Biol. 2021, 2, 12–29. [Google Scholar] [CrossRef]
- Zhong, X.; Wang, H.; Zhang, J.; Liu, H.; Zhang, S.; Song, H.-F.; Yang, G.; Zhang, L.; Ma, Y. Tellurium hydrides at high pressures: High-temperature superconductors. Phys. Rev. Lett. 2016, 116, 057002. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhang, Y.; You, Q.; Huang, P.; Wang, Y.; Huang, Z.N.; Ge, Y.; Wu, L.; Dong, Z.; Dai, X. Enhanced photodetection properties of tellurium@ selenium roll-to-roll nanotube heterojunctions. Small 2019, 15, 1900902. [Google Scholar] [CrossRef]
- Wang, L.; Cao, W.; Xu, H. Tellurium-containing polymers: Towards biomaterials and optoelectronic materials. ChemNanoMat 2016, 2, 479–488. [Google Scholar] [CrossRef]
- Steinke, T.; Wonner, P.; Engelage, E.; Huber, S.M. Catalytic Activation of a Carbon–Chloride Bond by Dicationic Tellurium-Based Chalcogen Bond Donors. Synthesis 2021, 53, 2043–2050. [Google Scholar]
- Sacramento, M.; Costa, G.P.; Barcellos, A.M.; Perin, G.; Lenardão, E.J.; Alves, D. Transition-Metal-Free C− S, C− Se, and C− Te Bond Formation from Organoboron Compounds. Chem. Rec. 2021, 21, 2855–2879. [Google Scholar] [CrossRef]
- Singh, A.; Kaushik, A.; Dhau, J.S.; Kumar, R. Exploring coordination preferences and biological applications of pyridyl-based organochalcogen (Se, Te) ligands. Coord. Chem. Rev. 2022, 450, 214254. [Google Scholar] [CrossRef]
- Ali, A.; Banerjee, B.; Srivastava, V.; Verma, V.K. Organochalcogen (Se/Te) substituted Schiff bases: Syntheses and applications. Mater. Today Proc. 2023, in press. [Google Scholar] [CrossRef]
- Zhou, J.; Qian, S.; Hao, B.; Liu, J.; Zhou, X.; Yan, C.; Qian, T. Small Molecules, Great Powers: Chemistry of Small Organo-Chalcogenide Molecules in Rechargeable Li-Sulfur Batteries. Adv. Funct. Mater. 2023, 2213966. [Google Scholar] [CrossRef]
- Hassan, A.F.; Abdalwahed, A.T.; Al-Luaibi, M.Y.; Aljadaan, S.A. Synthesis, Characterization and Thermal Study of some new Organochalcogenide compounds containing arylamide group. Egypt. J. Chem. 2021, 64, 5009–5015. [Google Scholar] [CrossRef]
- Kershaw, S.V.; Yiu, W.K.; Sergeev, A.; Rogach, A.L. Development of synthetic methods to grow long-wavelength infrared-emitting HgTe quantum dots in dimethylformamide. Chem. Mater. 2020, 32, 3930–3943. [Google Scholar] [CrossRef]
- Patsha, A.; Ranganathan, K.; Kazes, M.; Oron, D.; Ismach, A. Halide chemical vapor deposition of 2D semiconducting atomically-thin crystals: From self-seeded to epitaxial growth. Appl. Mater. Today 2022, 26, 101379. [Google Scholar] [CrossRef]
- Mane, R.; Mali, S.; Ghanwat, V.; Kondalkar, V.; Khot, K.; Mane, S.; Shinde, D.; Patil, P.; Bhosale, P. Photoelectrochemical Performance of MoBiInSe5 Mixed Metal Chalcogenide Thin Films. Mater. Today Proc. 2015, 2, 1458–1463. [Google Scholar] [CrossRef]
- Yeo, J.S.; Vittal, J.J.; Henderson, W.; Hor, T.A. Ligand functionalization, reactivity, and transformation at the selenide centers of [Pt2 (μ-Se) 2 (PPh3) 4] with organic halides. Organometallics 2002, 21, 2944–2949. [Google Scholar] [CrossRef]
- Frogley, B.J.; Hill, A.F.; Watson, L.J. Bridging selenocarbonyl ligands: An open and shut case. Chem. Commun. 2019, 55, 14450–14453. [Google Scholar] [CrossRef]
- Hesford, M.; Levason, W.; Orchard, S.D.; Reid, G. Synthesis and complexation of a new facultative tridentate S2Te donor ligand MeS (CH2) 3Te (CH2) 3SMe: Crystal structures of [Rh (Cp*)(S2Te)][PF6] 2 and [PtCl (S2Te)] PF6. J. Organomet. Chem. 2002, 649, 214–218. [Google Scholar] [CrossRef]
- Kieser, J.M.; Jones, L.O.; Uible, M.C.; Zeller, M.; Schatz, G.C.; Bart, S.C. Late to the Party: Synthesis and Characterization of Tellurium and Selenium Half-Sandwich Complexes. Organometallics 2021, 40, 4104–4109. [Google Scholar] [CrossRef]
- Molonia, M.S.; Muscarà, C.; Speciale, A.; Salamone, F.L.; Toscano, G.; Saija, A.; Cimino, F. The p-Phthalates Terephthalic Acid and Dimethyl Terephthalate Used in the Manufacture of PET Induce In Vitro Adipocytes Dysfunction by Altering Adipogenesis and Thermogenesis Mechanisms. Molecules 2022, 27, 7645. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Rusakova, I.L. Long-range relativistic heavy atom effect on 1H NMR chemical shifts of selenium-and tellurium-containing compounds. Int. J. Quantum Chem. 2019, 119, e25809. [Google Scholar] [CrossRef]
- Pirillo, J.; De Simone, B.C.; Russo, N. Photophysical properties prediction of selenium-and tellurium-substituted thymidine as potential UVA chemotherapeutic agents. Theor. Chem. Acc. 2016, 135, 1–5. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Rusakova, I.L. Hierarchical basis sets for the calculation of nuclear magnetic resonance spin–spin coupling constants involving either selenium or tellurium nuclei. J. Phys. Chem. A 2019, 123, 6564–6571. [Google Scholar] [CrossRef]
- Roca Jungfer, M.; Schulz Lang, E.; Abram, U. Solvents and Ligands Matter: Structurally Variable Palladium and Nickel Clusters Assembled by Tridentate Selenium-and Tellurium-Containing Schiff Bases. Inorg. Chem. 2022, 61, 3785–3800. [Google Scholar] [CrossRef]
- Sharma, T.; Sharma, R.; Tamboli, R.A.; Kanhere, D.G. Ab initio investigation of structural and electronic properties of selenium and tellurium clusters. Eur. Phys. J. B 2019, 92, 1–14. [Google Scholar] [CrossRef]
- Nagashima, Y.; Ishigaki, S.; Tanaka, J.; Tanaka, K. Acceleration Mechanisms of C–H Bond Functionalization Catalyzed by Electron-Deficient CpRh (III) Complexes. ACS Catal. 2021, 11, 13591–13602. [Google Scholar] [CrossRef]
- Zakharov, A.V.; Sadekov, I.D.; Minkin, V.I. Synthesis, reactions and structures of telluronium salts. Russ. Chem. Rev. 2006, 75, 207. [Google Scholar] [CrossRef]
- Günther, W.; Nepywoda, J.; Chu, J. Methods in chalcogen chemistry: V. A new reagent for the synthesis of aromatic tellurium compounds. J. Organomet. Chem. 1974, 74, 79–84. [Google Scholar] [CrossRef]
- Abd-Elnaiem, A.M.; Abdelraheem, A.; Abdel-Rahim, M.; Moustafa, S. Substituting silver for tellurium in selenium–tellurium thin films for improving the optical characteristics. J. Inorg. Organomet. Polym. Mater. 2022, 32, 2009–2021. [Google Scholar] [CrossRef]
- Abu-Sehly, A.; Rashad, M.; Hafiz, M.; Abd-Elmageed, A.; Amin, R. Tuning optical properties of thin films based on selenium tellurium. Opt. Mater. 2020, 109, 110291. [Google Scholar] [CrossRef]
- Rashad, M.; Shaalan, N.; Abd-Elmageed, A.; Amin, R.; Hafiz, M.; Abu-Sehly, A. The effects of different dopant on the optical parameters for selenium tellurium thin films. Optik 2021, 241, 166102. [Google Scholar] [CrossRef]
- Hadar, I.; Hu, X.; Luo, Z.-Z.; Dravid, V.P.; Kanatzidis, M.G. Nonlinear band gap tunability in selenium–tellurium alloys and its utilization in solar cells. ACS Energy Lett. 2019, 4, 2137–2143. [Google Scholar] [CrossRef]
- Mondal, R.; Biswas, D.; Paul, S.; Das, A.S.; Chakrabarti, C.; Roy, D.; Bhattacharya, S.; Kabi, S. Investigation of microstructural, optical, physical properties and dielectric relaxation process of sulphur incorporated selenium–tellurium ternary glassy systems. Mater. Chem. Phys. 2021, 257, 123793. [Google Scholar] [CrossRef]
- Barca, G.M.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Salavati-Niasari, M.; Mirsattari, S.; Monajjemi, M.; Hamadanian, M. Density functional B3LYP and B3PW91 studies of the properties of four cyclic organodiboranes with tetramethylene fragments. J. Struct. Chem. 2010, 51, 437–443. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functional theory for reaction energies: Test of meta and hybrid meta functionals, range-separated functionals, and other high-performance functionals. J. Chem. Theory Comput. 2011, 7, 669–676. [Google Scholar] [CrossRef]
- Kossmann, S.; Kirchner, B.; Neese, F. Performance of modern density functional theory for the prediction of hyperfine structure: Meta-GGA and double hybrid functionals. Mol. Phys. 2007, 105, 2049–2071. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Verma, P.; Jin, X.; Truhlar, D.G.; He, X. Revised M06 density functional for main-group and transition-metal chemistry. Proc. Natl. Acad. Sci. USA 2018, 115, 10257–10262. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the performance of the M05− 2X and M06− 2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Iyer, S.; Lopez-Hilfiker, F.; Lee, B.H.; Thornton, J.A.; Kurten, T. Modeling the detection of organic and inorganic compounds using iodide-based chemical ionization. J. Phys. Chem. A 2016, 120, 576–587. [Google Scholar] [CrossRef]
- Karton, A. How reliable is DFT in predicting relative energies of polycyclic aromatic hydrocarbon isomers? comparison of functionals from different rungs of jacob’s ladder. J. Comput. Chem. 2017, 38, 370–382. [Google Scholar] [CrossRef]
- Nakajima, Y.; Seino, J.; Nakai, H. Relativistic effect on enthalpy of formation for transition-metal complexes. Chem. Phys. Lett. 2017, 673, 24–29. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* Basis Set for Third-Row Atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Xu, X.; Truhlar, D.G. Accuracy of effective core potentials and basis sets for density functional calculations, including relativistic effects, as illustrated by calculations on arsenic compounds. J. Chem. Theory Comput. 2011, 7, 2766–2779. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Prakash, R.; Nandi, C.; Chowdhury, M.G.; Raghavendra, B.; Roisnel, T.; Ghosh, S. Syntheses and structures of chalcogen-bridged binuclear group 5 and 6 metal complexes. J. Chem. Sci. 2019, 131, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Seino, H.; Mizobe, Y. Reactions of Bis (chalcogenolato) Complexes [(η5-C5Me5) Ir (CO)(ETol) 2](E= Se, S; Tol= p-Tolyl) with [Pt (PPh3) 3]. Formation of Tri-or Dinuclear Mixed-Metal Complexes with Bridging Chalcogenido or Chalcogenolato Ligands. Organometallics 2010, 29, 2254–2259. [Google Scholar] [CrossRef]
- Goerigk, L. A Comprehensive Overview of the DFT-D3 London-Dispersion Correction. In Non-Covalent Interactions in Quantum Chemistry and Physics; Elsevier: Cambridge, UK, 2017; pp. 195–219. [Google Scholar]
- Smith, D.G.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Hoggan, P.E. Proceedings of MEST 2012: Electronic Structure Methods with Applications to Experimental Chemistry; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Liu, W.-K.; Kong, S.-S.; Yu, X.-X.; Li, Y.-L.; Yang, L.-Z.; Ma, Y.; Fang, X.-Y. Interlayer coupling, electronic and optical properties of few-layer silicon carbide nanosheets. Mater. Today Commun. 2023, 34, 105030. [Google Scholar] [CrossRef]
- Kong, S.-S.; Liu, W.-K.; Yu, X.-X.; Li, Y.-L.; Yang, L.-Z.; Ma, Y.; Fang, X.-Y. Interlayer interaction mechanism and its regulation on optical properties of bilayer SiCNSs. Front. Phys. 2023, 18, 43302. [Google Scholar] [CrossRef]
- Hao, P.; Sun, J.; Xiao, B.; Ruzsinszky, A.; Csonka, G.I.; Tao, J.; Glindmeyer, S.; Perdew, J.P. Performance of meta-GGA functionals on general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2013, 9, 355–363. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, X.; Yu, H.S.; Truhlar, D.G.; He, X. Revised M06-L functional for improved accuracy on chemical reaction barrier heights, noncovalent interactions, and solid-state physics. Proc. Natl. Acad. Sci. USA 2017, 114, 8487–8492. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-X.; Chen, W.; Yan, X.; Xie, F.; Wei, J.; Ye, S.; Xi, Z. Preparation and Characterization of a 19-Electron Ni (I) Complex Bearing a Cyclopentadienyl-Phosphine Ligand. Organometallics 2022, 41, 2227–2231. [Google Scholar] [CrossRef]
- Silva, T.J.; Mendes, P.J.; Santos, A.M.; Garcia, M.H.; Robalo, M.P.; Ramalho, J.P.; Carvalho, A.P.; Büchert, M.; Wittenburg, C.; Heck, J.r. Mono (η5-cyclopentadienyl) metal (II) complexes with thienyl acetylide chromophores: Synthesis, electrochemical studies, and first hyperpolarizabilities. Organometallics 2014, 33, 4655–4671. [Google Scholar] [CrossRef]
- Bondi, A.v. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Gaus, P.L. Basic Inorganic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 1995. [Google Scholar]
- Miar, M.; Shiroudi, A.; Pourshamsian, K.; Oliaey, A.R.; Hatamjafari, F. Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. J. Chem. Res. 2021, 45, 147–158. [Google Scholar]
- Li, G.; Chen, X.; Zhou, Z.; Wang, F.; Yang, H.; Yang, J.; Xu, B.; Yang, B.; Liu, D. Theoretical insights into the structural, relative stable, electronic, and gas sensing properties of Pb n Au n (n= 2–12) clusters: A DFT study. RSC Adv. 2017, 7, 45432–45441. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Shi, W.; Zhu, Y. Internal electric field engineering for steering photogenerated charge separation and enhancing photoactivity. EcoMat 2019, 1, e12007. [Google Scholar] [CrossRef] [Green Version]
- Coehoorn, R.; Lin, X.; Weijtens, C.; Gottardi, S.; Van Eersel, H. Three-Dimensional Modeling of Organic Light-Emitting Diodes Containing Molecules with Large Electric Dipole Moments. Phys. Rev. Appl. 2021, 16, 034048. [Google Scholar] [CrossRef]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Rahman, F.-U.; Yu, Y.; Rebek, J. The role of electric field, peripheral chains, and magnetic effects on significant 1 H upfield shifts of the encapsulated molecules in chalcogen-bonded capsules. Phys. Chem. Chem. Phys. 2021, 23, 19647–19658. [Google Scholar] [CrossRef]
- Thomas, A.A.; Speck, K.; Kevlishvili, I.; Lu, Z.; Liu, P.; Buchwald, S.L. Mechanistically guided design of ligands that significantly improve the efficiency of CuH-catalyzed hydroamination reactions. J. Am. Chem. Soc. 2018, 140, 13976–13984. [Google Scholar] [CrossRef]
- Cui, C.-H.; Yu, S.-H. Engineering interface and surface of noble metal nanoparticle nanotubes toward enhanced catalytic activity for fuel cell applications. Acc. Chem. Res. 2013, 46, 1427–1437. [Google Scholar] [CrossRef]
- Li, H.H.; Yu, S.H. Recent advances on controlled synthesis and engineering of hollow alloyed nanotubes for electrocatalysis. Adv. Mater. 2019, 31, 1803503. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Gong, P.; Ma, W.-D.; Hao, R.; Fang, X.-Y. Effects of substitution of group-V atoms for carbon or silicon atoms on optical properties of silicon carbide nanotubes. Chin. Phys. B 2021, 30, 067803. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid GGA | Meta-GGA | Hybrid–Meta-GGA | |||||
|---|---|---|---|---|---|---|---|
| HOMO–LUMO gap(eV)/complex | B3LYP | PBE0 | B3PW91 | M06-L | TPSS | M06 | M06-2X |
| [Cp][(TePh3)] | 3.631 | 4.187 | 3.741 | 2.640 | 2.649 | 4.136 | 5.841 |
| [Cp][(SePh3)] | 3.317 | 3.784 | 3.447 | 2.189 | 2.366 | 3.687 | 5.364 |
| Complexes | Distance, C(1)~C(5) | Distance, E-[C(1)~C(5)] | Distance, E-Ci~iii | Distance, E-c5 |
|---|---|---|---|---|
| [Cp][(TePh3)] (1) | 1.412–1.434 | 2.697–3.209 | 2.162–2.211 | 2.739 |
| [CpMe4][(TePh3)] (2) | 1.413–1.438 | 2.733–3.123 | 2.169–2.218 | 2.689 |
| [CpMe5][(TePh3)] (3) | 1.417–1.440 | 2.764–3.139 | 2.157–2.216 | 2.702 |
| [Cp][(SePh3)] (4) | 1.417–1.425 | 2.854–2.915 | 1.982–1.989 | 2.602 |
| [CpMe5][(SePh3)] (5) | 1.424–1.431 | 2.779–2.948 | 1.969–2.009 | 2.615 |
| ∠c5-E-ci | ∠c5-E-cii | ∠c5-E-ciii | ∠c5-E-c3 | |
| [Cp][(TePh3)] (1) | 153.127 | 108.085 | 108.858 | 177.13 |
| [CpMe4][(TePh3)] (2) | 148.607 | 109.875 | 112.531 | 166.76 |
| [CpMe5][(TePh3)] (3) | 146.07 | 106.45 | 119.33 | 169.45 |
| [Cp][(SePh3)] (4) | 124.74 | 118.95 | 120.53 | 177.36 |
| [CpMe5][(SePh3)] (5) | 126.393 | 113.006 | 125.806 | 174.33 |
| ∠ci-E-cii | ∠ci-E-ciii | ∠cii-E-ciii | ||
| [Cp][(TePh3)] (1) | 90.418 | 89.209 | 92.022 | |
| [CpMe4][(TePh3)] (2) | 88.265 | 91.821 | 90.940 | |
| [CpMe5][(TePh3)] (3) | 89.447 | 89.604 | 90.310 | |
| [Cp][(SePh3)] (4) | 95.03 | 94.80 | 95.94 | |
| [CpMe5][(SePh3)] (5) | 95.466 | 92.440 | 96.429 |
| Complexes | ΔE | η5-Distance | Eg | IP | EA | Dipole Moment |
|---|---|---|---|---|---|---|
| [Cp][(TePh3)] | −59.93 | 2.74 | 2.65 | 4.27 | 1.62 | 4.74 |
| [CpMe4][(TePh3)] | −55.95 | 2.69 | 2.42 | 3.96 | 1.54 | 3.71 |
| [CpMe5][(TePh3)] | −55.17 | 2.70 | 2.25 | 3.84 | 1.56 | 3.44 |
| [Cp][(SePh3)] | −62.90 | 2.60 | 2.37 | 3.93 | 1.56 | 6.23 |
| [CpMe5][(SePh3)] | −58.16 | 2.62 | 1.98 | 3.55 | 1.57 | 4.92 |
| Complexes | HOMO | LUMO |
|---|---|---|
| [Cp][(TePh3)] |  |  |
| [CpMe4][(TePh3)] |  |  |
| [CpMe5][(TePh3)] |  |  |
| [Cp][(SePh3)] |  |  |
| [CpMe5][(SePh3)] |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyeniyi, G.T.; Melchakova, I.A.; Polyutov, S.P.; Avramov, P.V. Theoretical DFT Investigation of Structure and Electronic Properties of η5-Cyclopentadienyl Half-Sandwich Organochalcogenide Complexes. Electronics 2023, 12, 2738. https://doi.org/10.3390/electronics12122738
Oyeniyi GT, Melchakova IA, Polyutov SP, Avramov PV. Theoretical DFT Investigation of Structure and Electronic Properties of η5-Cyclopentadienyl Half-Sandwich Organochalcogenide Complexes. Electronics. 2023; 12(12):2738. https://doi.org/10.3390/electronics12122738
Chicago/Turabian StyleOyeniyi, G. T., Iu. A. Melchakova, S. P. Polyutov, and P. V. Avramov. 2023. "Theoretical DFT Investigation of Structure and Electronic Properties of η5-Cyclopentadienyl Half-Sandwich Organochalcogenide Complexes" Electronics 12, no. 12: 2738. https://doi.org/10.3390/electronics12122738