
Identification of Secondary Breast Cancer in Vital Organs through the Integration of Machine Learning and Microarrays

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Background
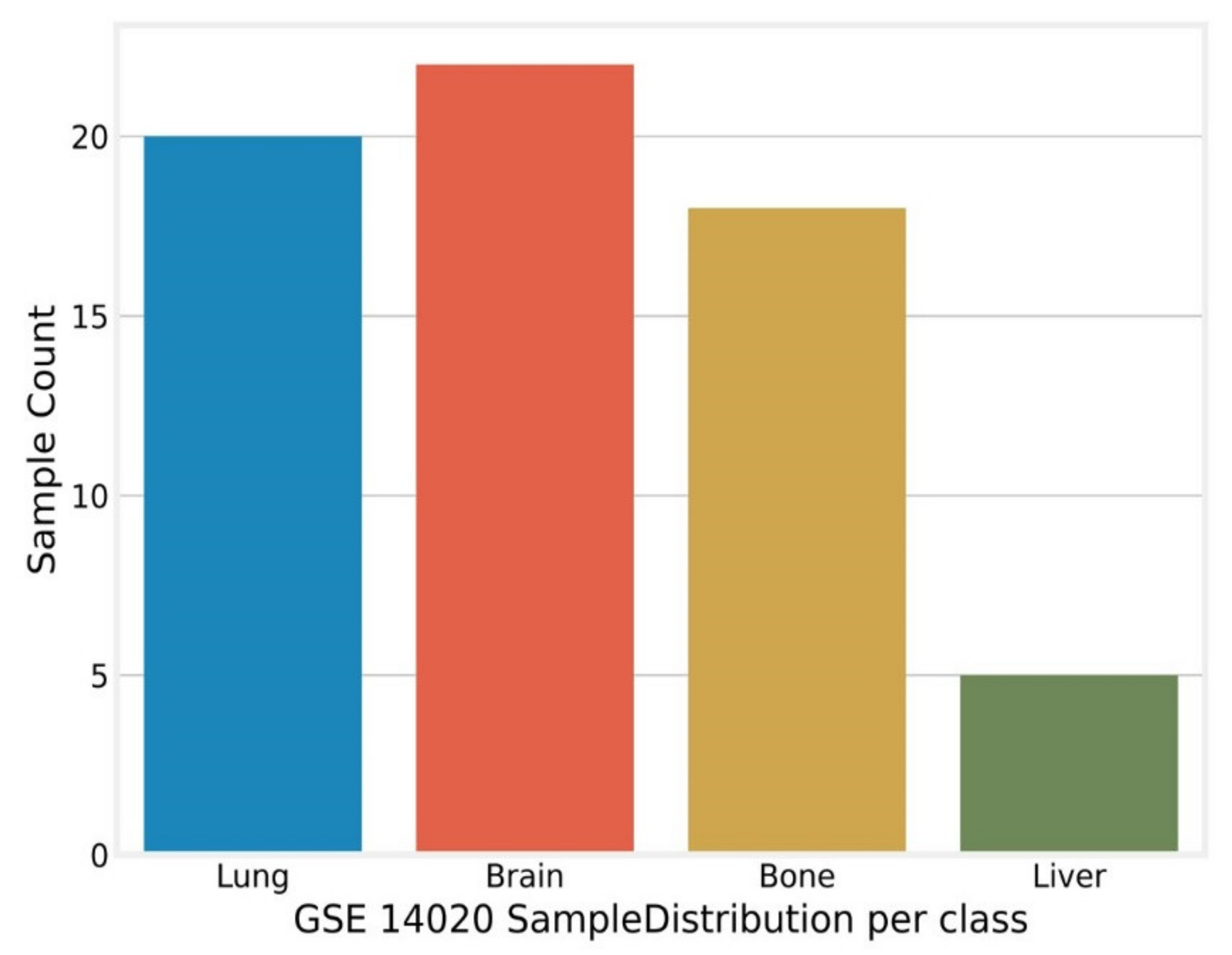
2.2. Breast Cancer Bone Metastasis (BCBoM)
2.3. Breast Cancer Liver Metastasis (BCLiM)
2.4. Breast Cancer Brain Metastasis (BCBrM)
2.5. Breast Cancer Lung Metastasis (BCLuM)
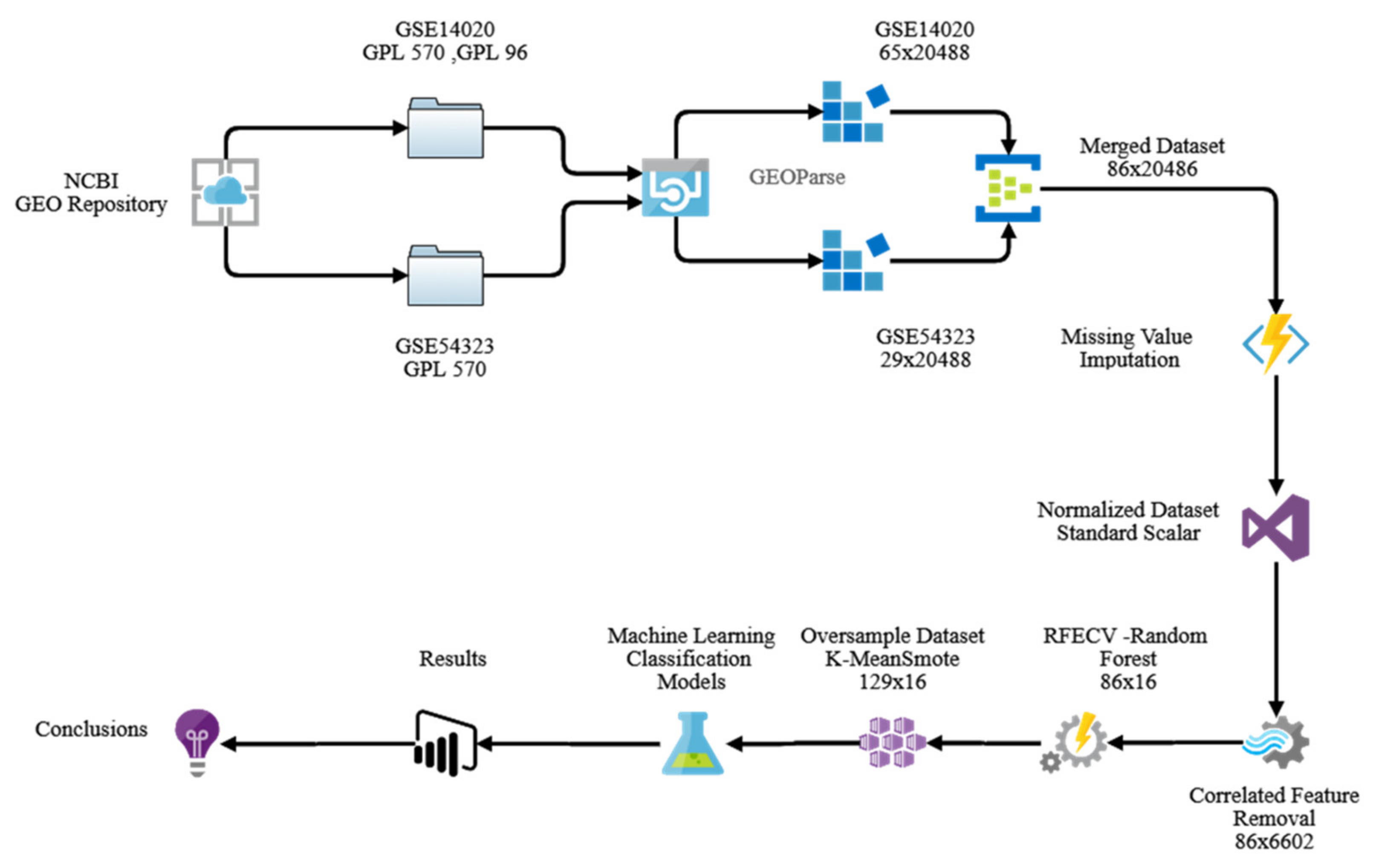
2.6. Methodology
2.7. Dataset (Dataset Availability)
- Entrez Gene
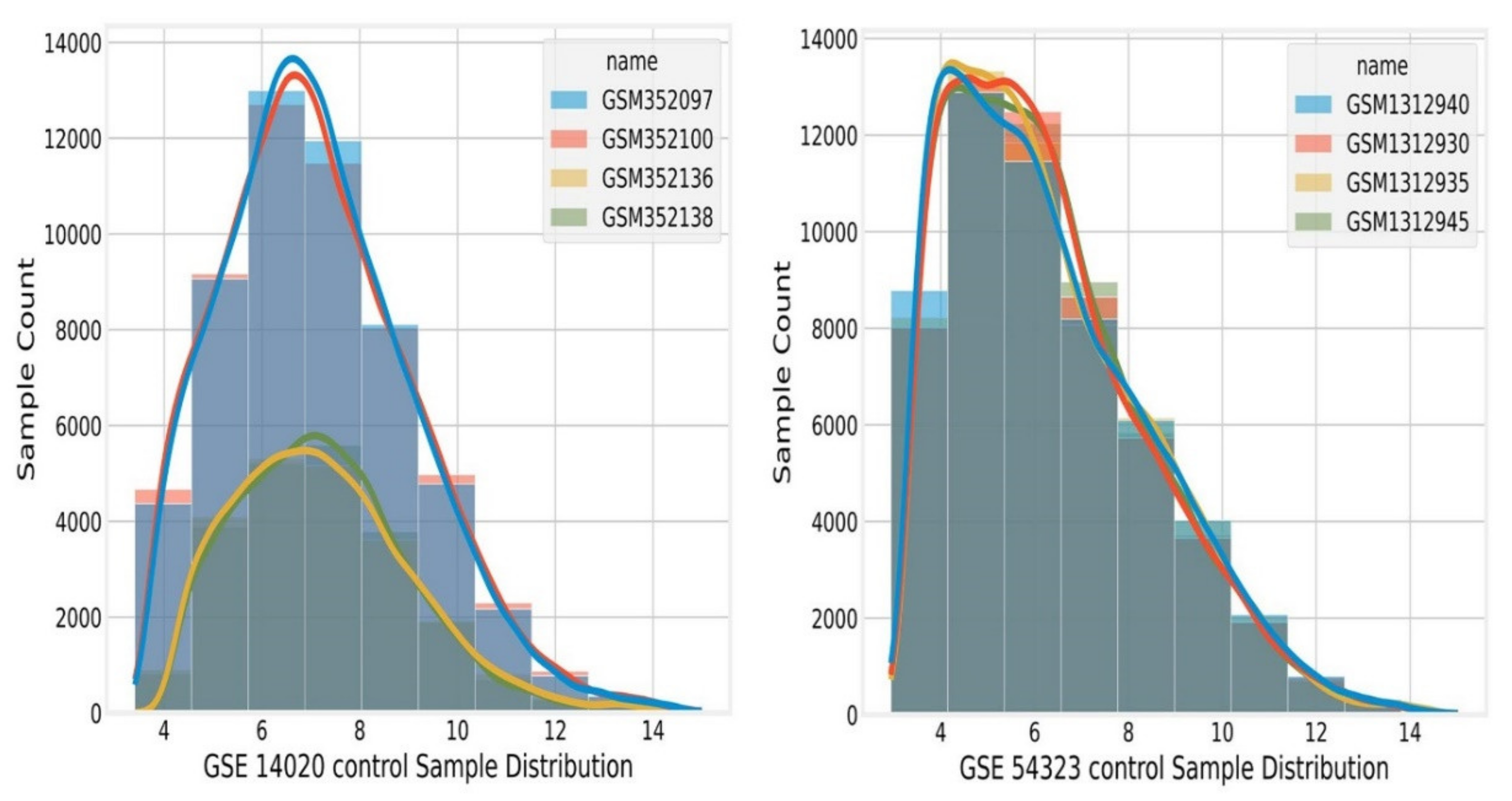
2.8. Data Pre-Processing
- SOFT Files
- GEOParse
- Download supplementary files for the GEO series to use locally.
- Load GEO SOFT as easy to use and manipulate objects.
- Prepare data for GEO upload.
2.9. Data Transformation
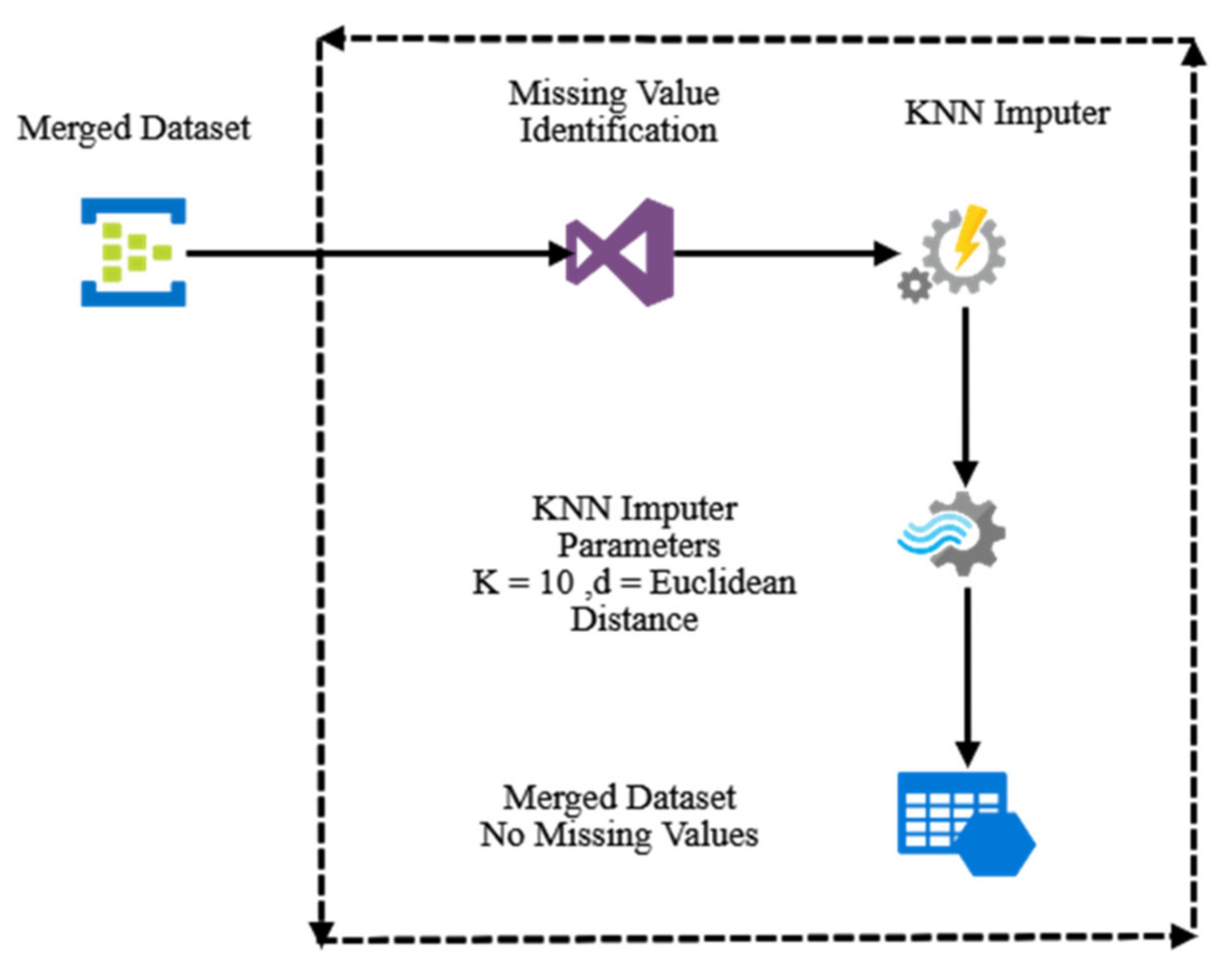
- Missing value imputation
- KNN (K-nearest neighbors) impute
- With a predefined value of K, the KNN is computed.
- With a predefined radius/threshold r, all the neighbors whose distance is less than or equal to the radius are computed.
- The KNN-based method
- Data Normalization
- Where mean: and standard deviation:
- Removal of Correlated Variable
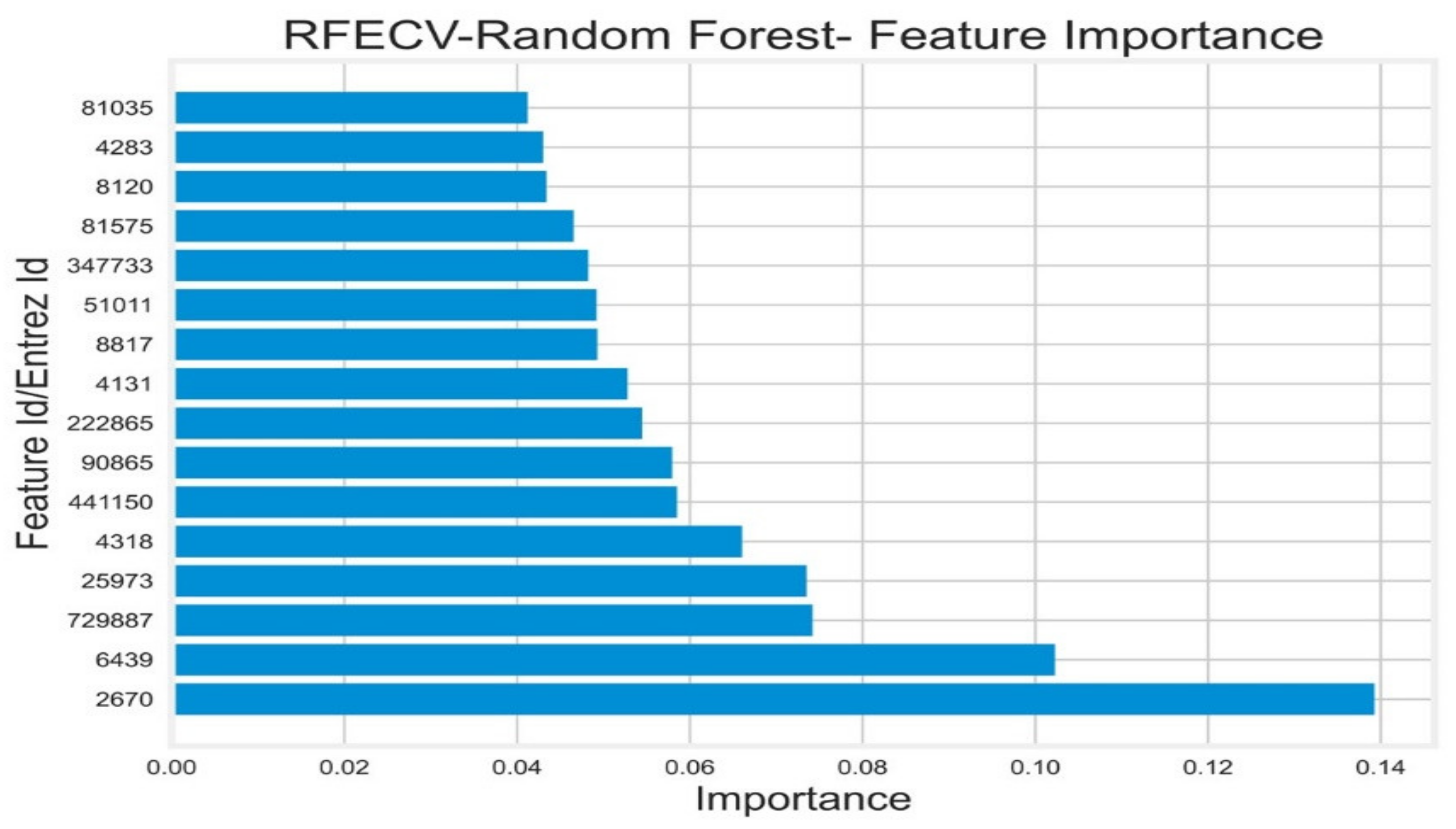
- Dimensionality Reduction
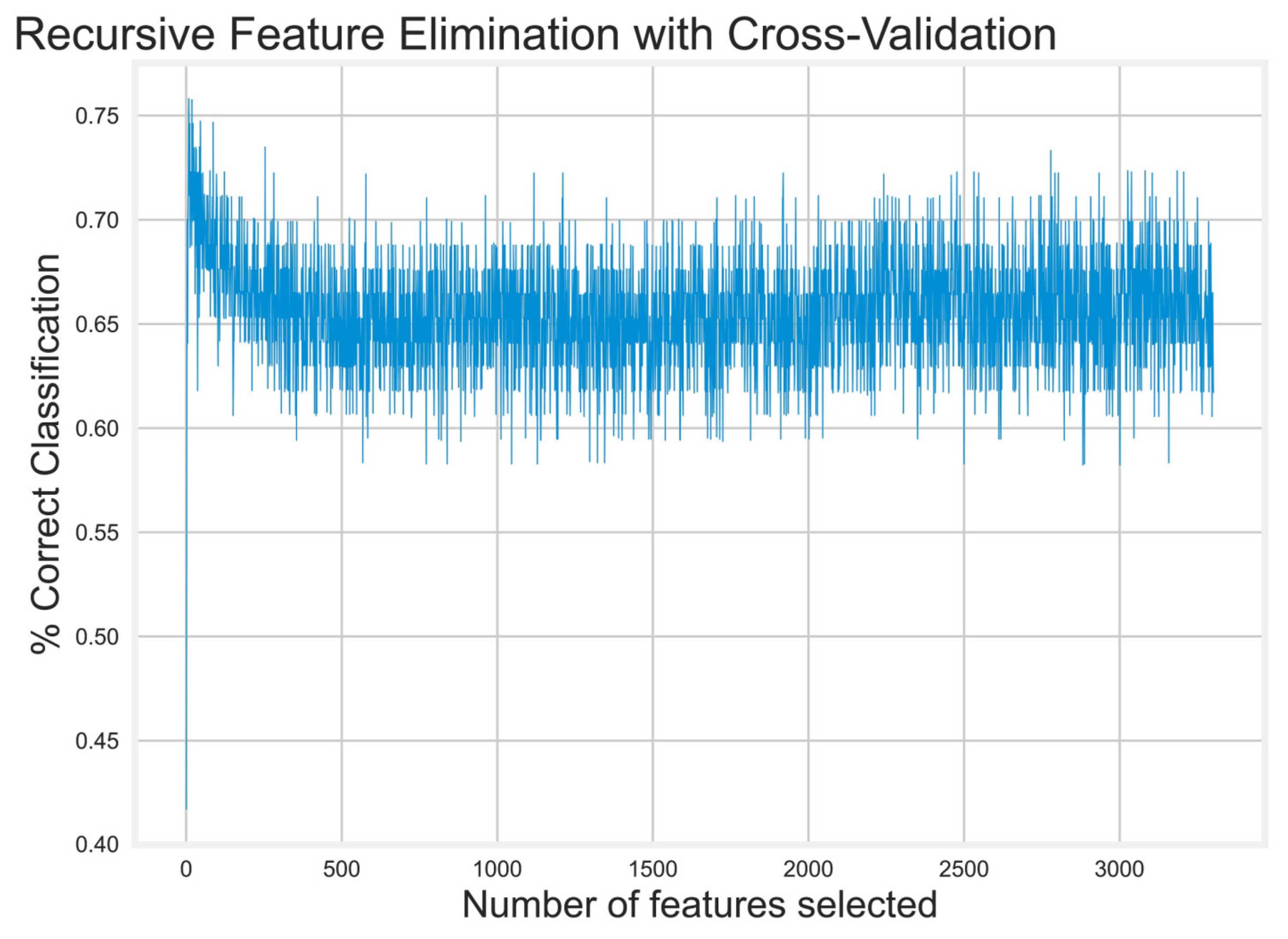
- Recursive Feature Elimination (RFE)
- Class imbalance
- SMOTE
- Use the K-means cluster algorithm to cluster entire data.
- Choose clusters with a significant number of minority class samples.
- Assign more synthetic samples to clusters with sparse distribution of minority class samples.
- Sampling
2.10. Classification Models
- K-nearest neighbor (KNN)
- Decision Trees (DTs)
- Random Forests (RF)
- Support Vector Machine (SVM)
2.11. Classification Evaluation Metrics
3. Results and Discussion
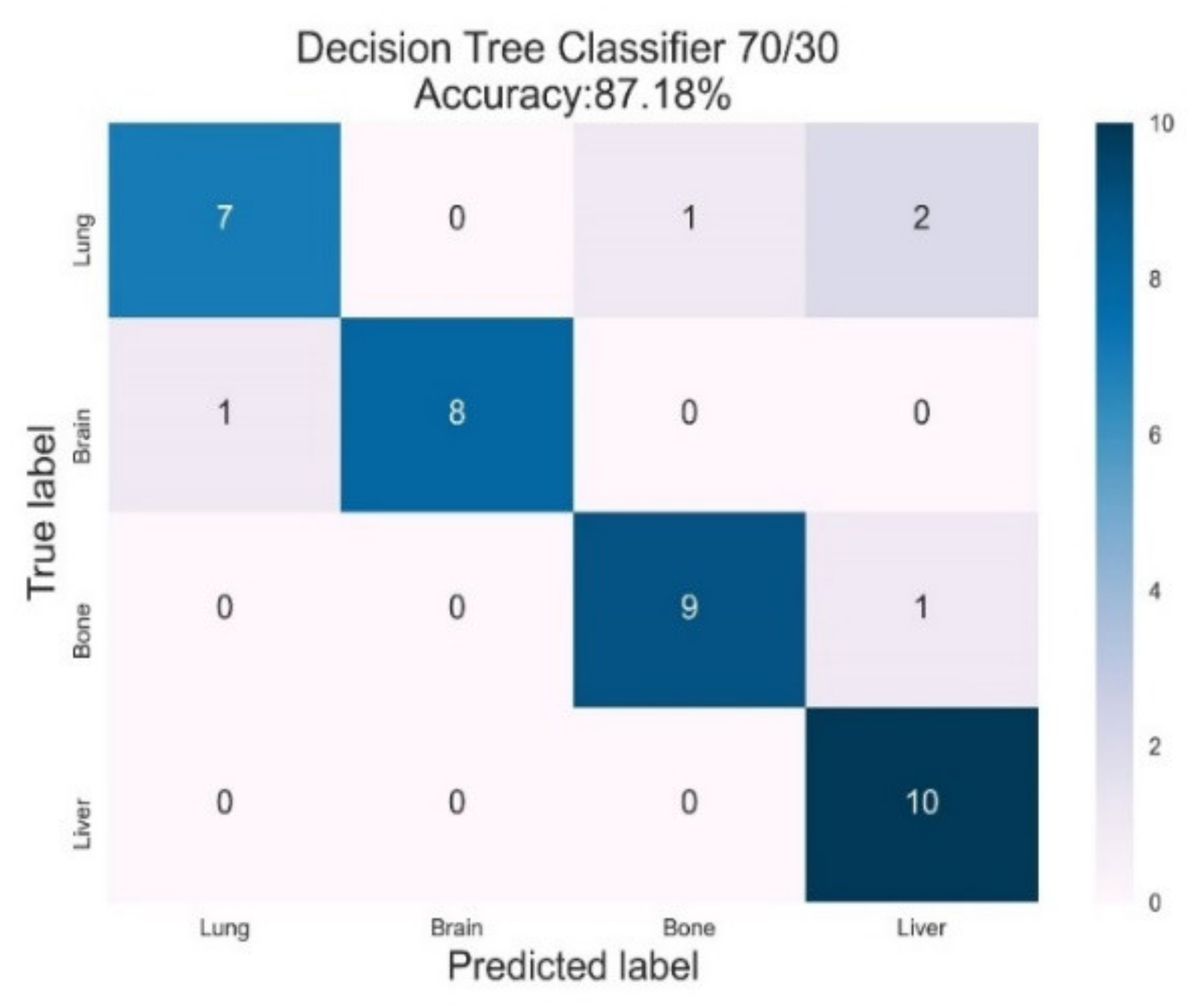
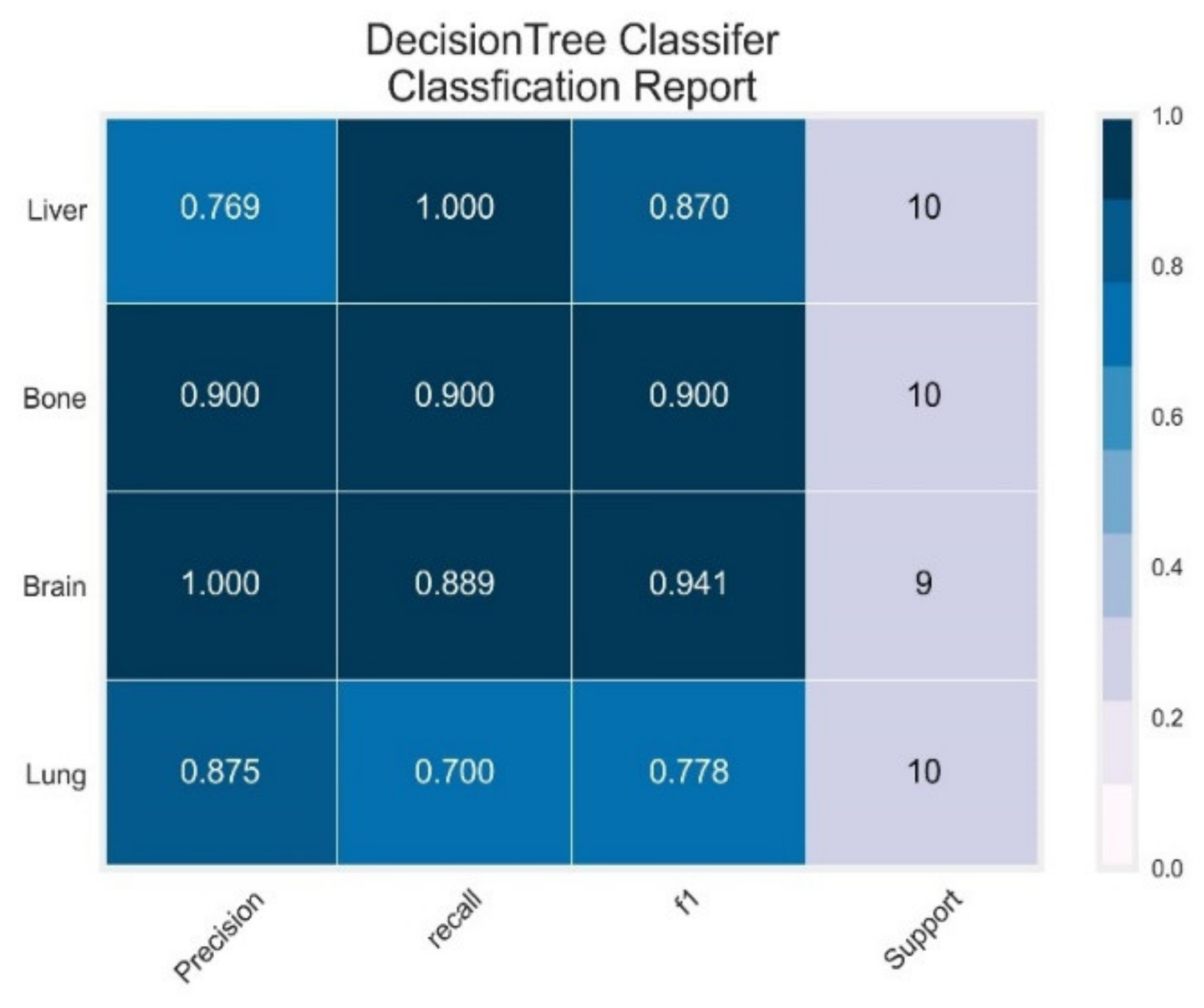
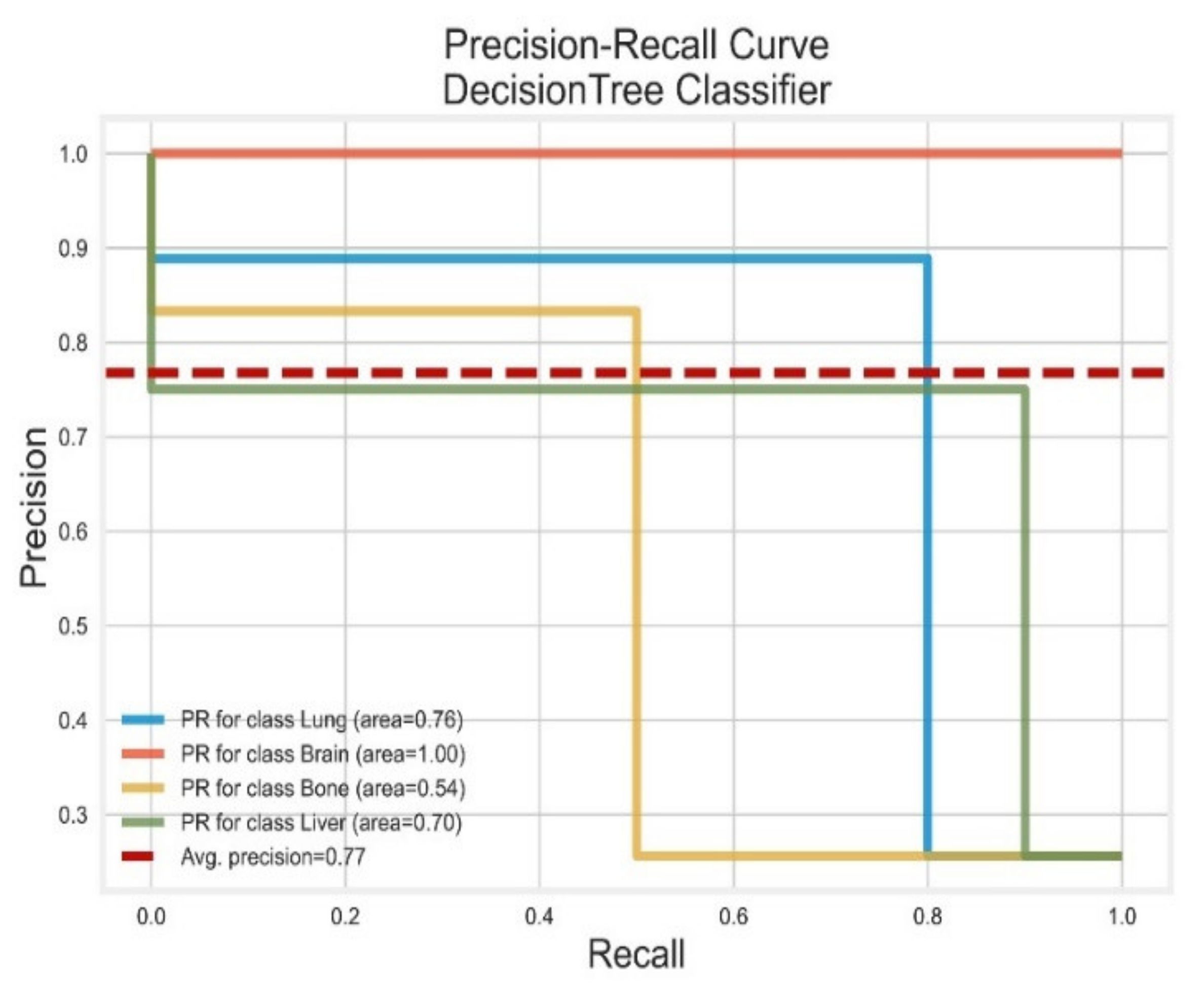
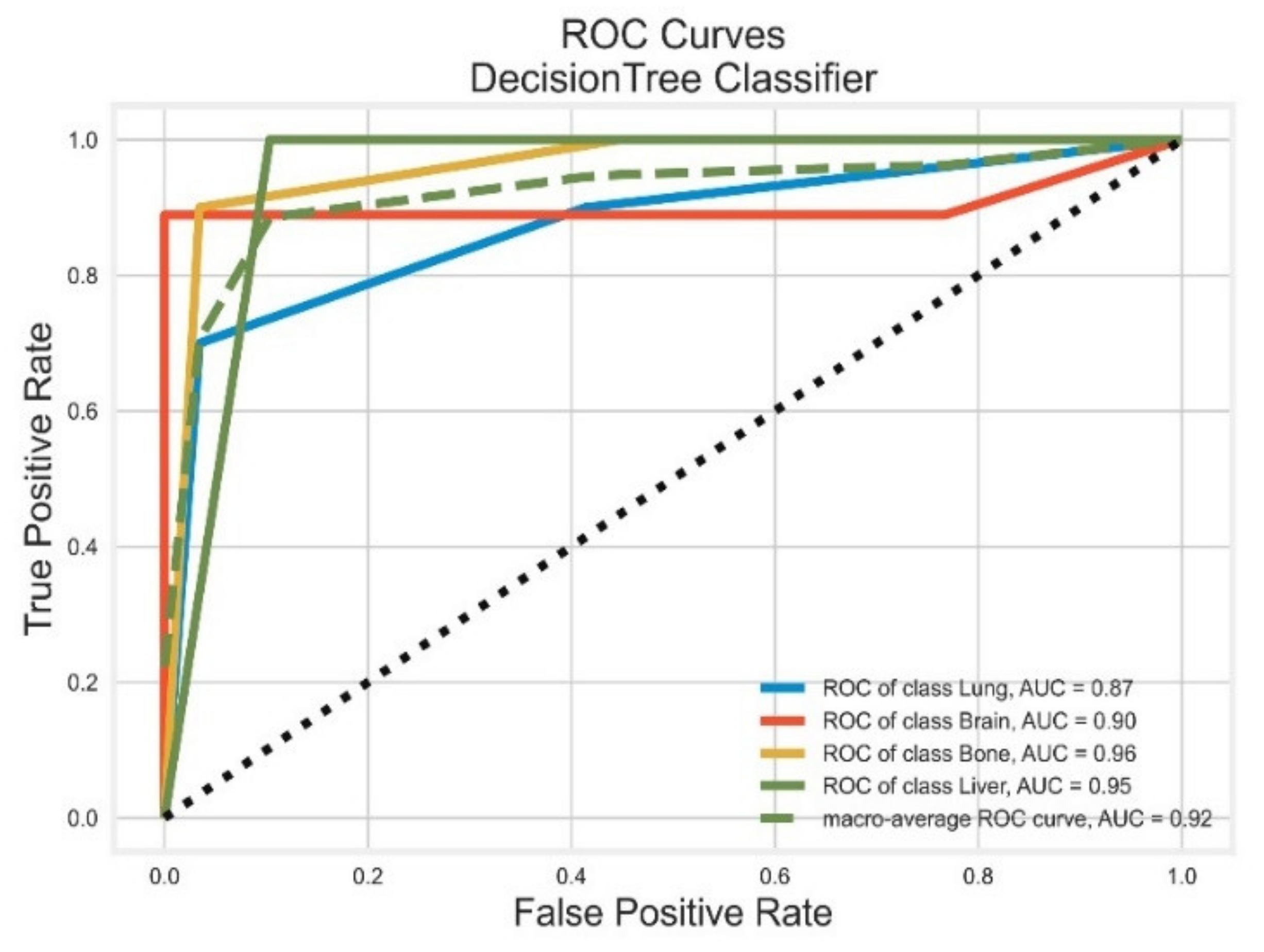
3.1. Decision Tree Classifier
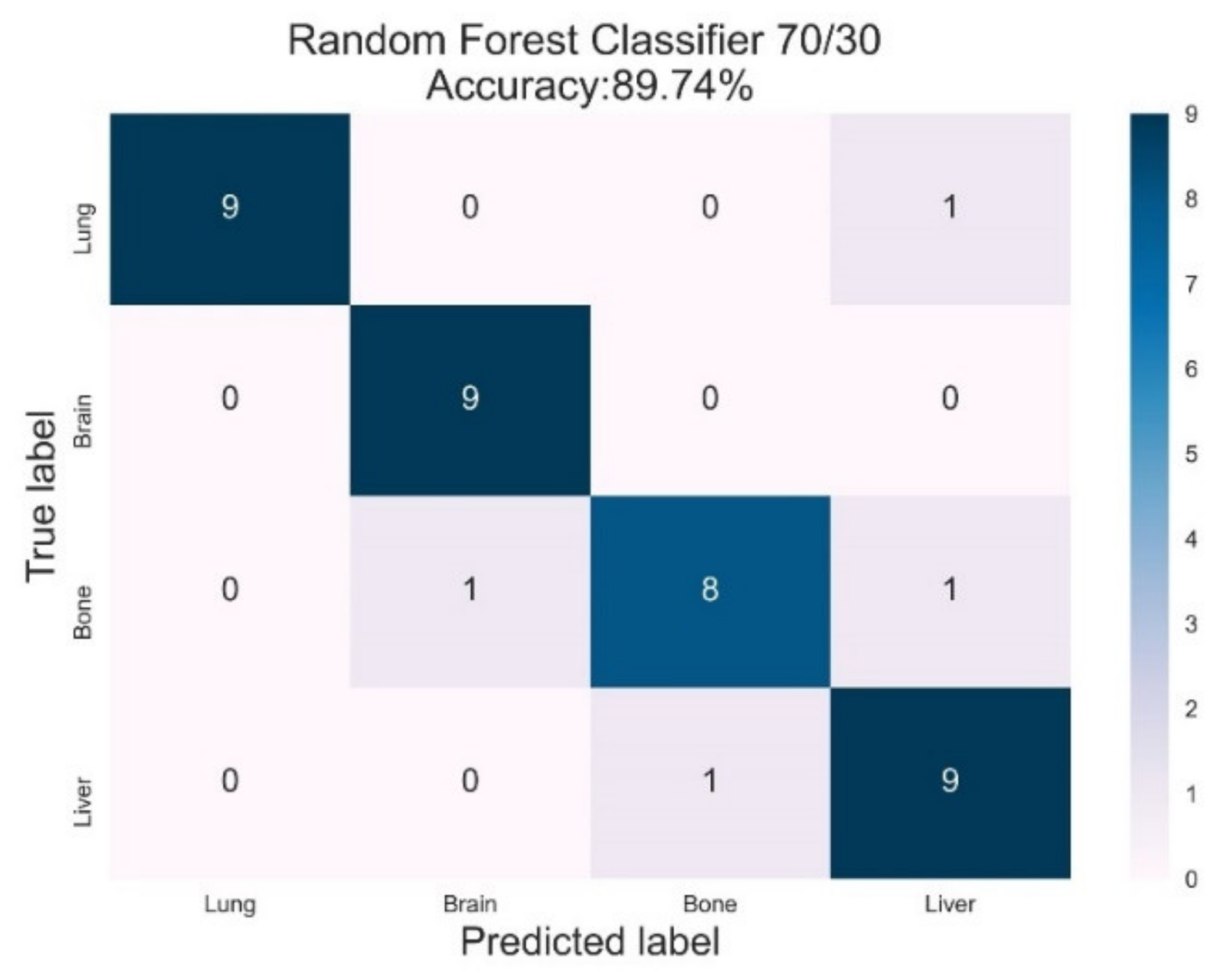
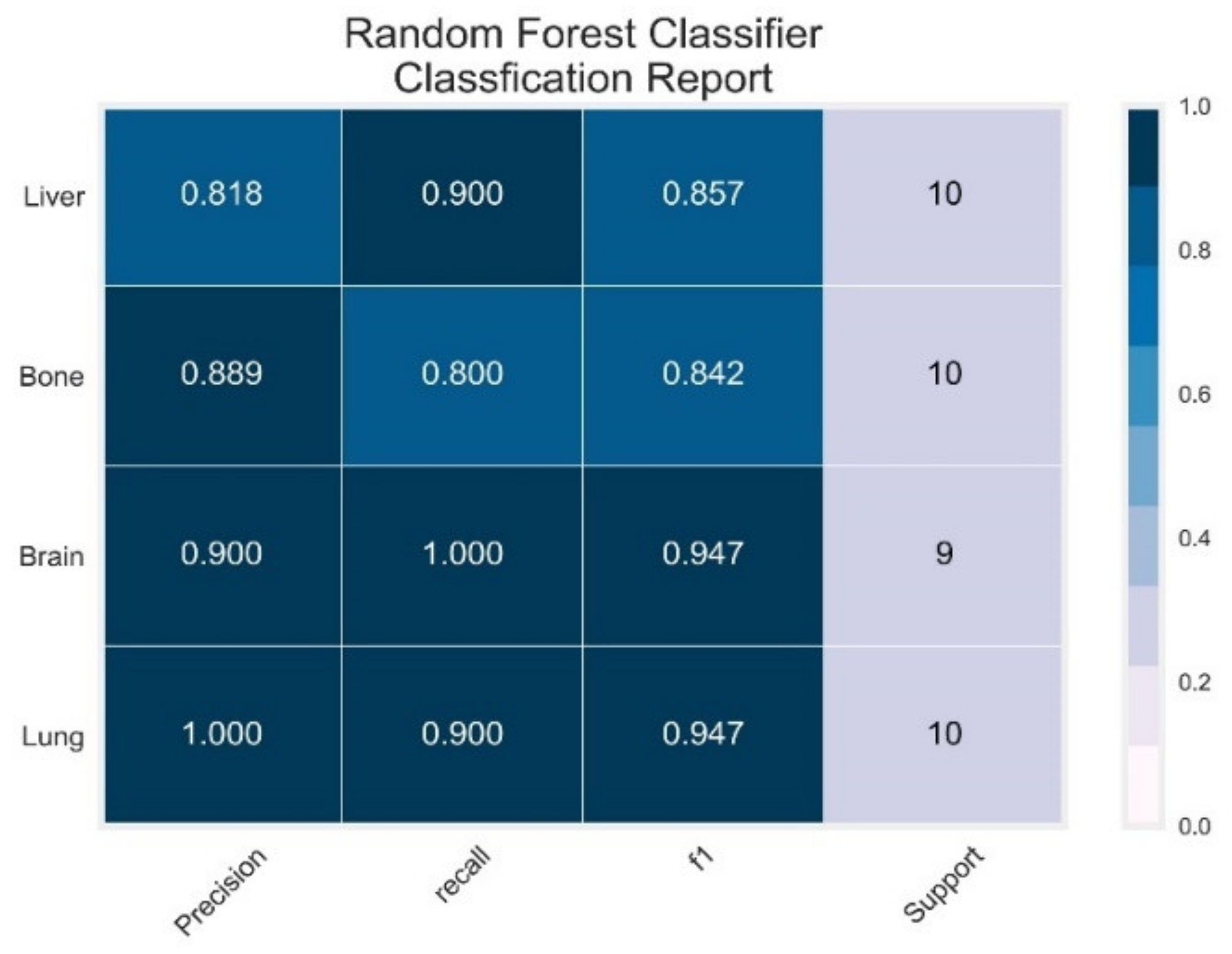
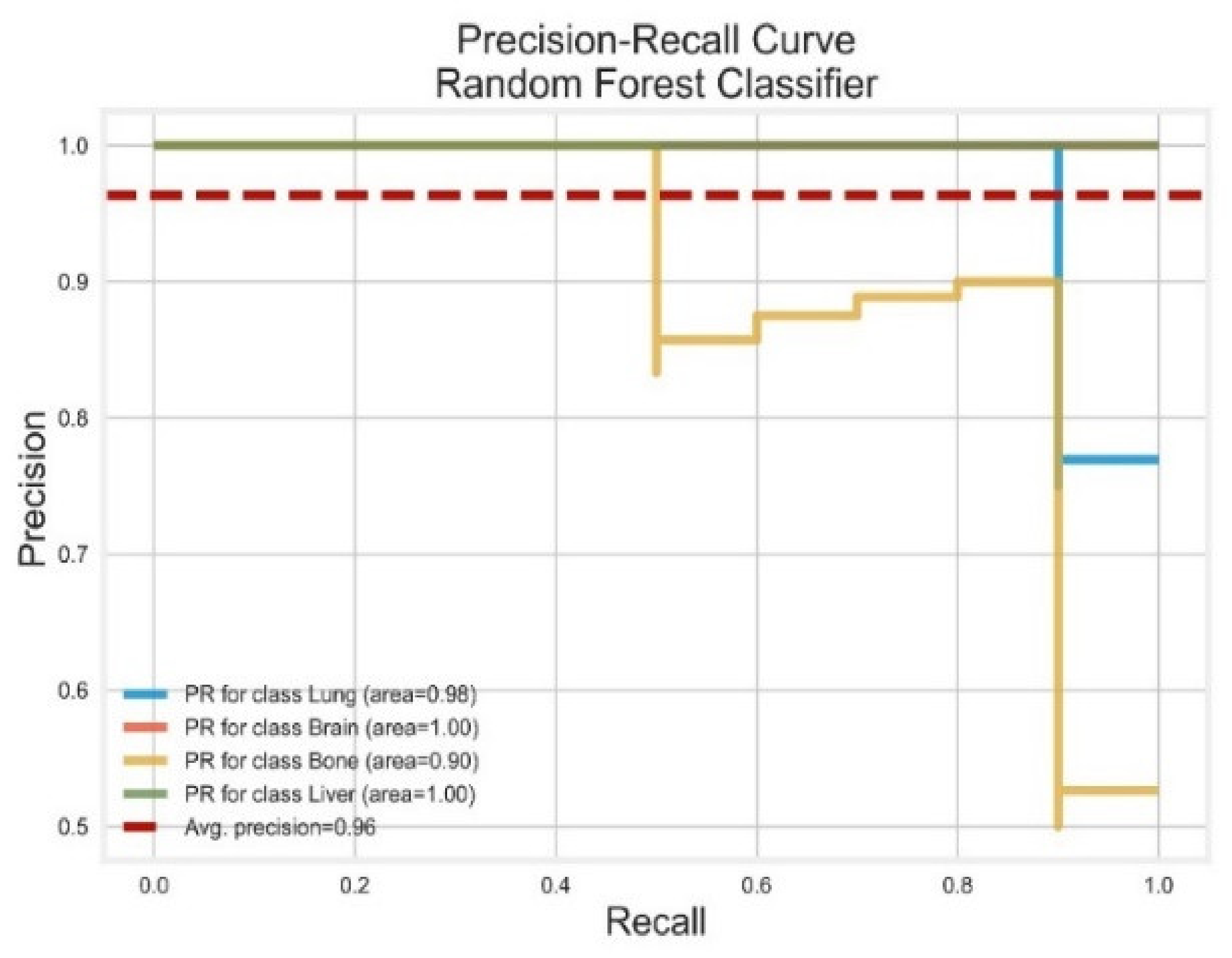
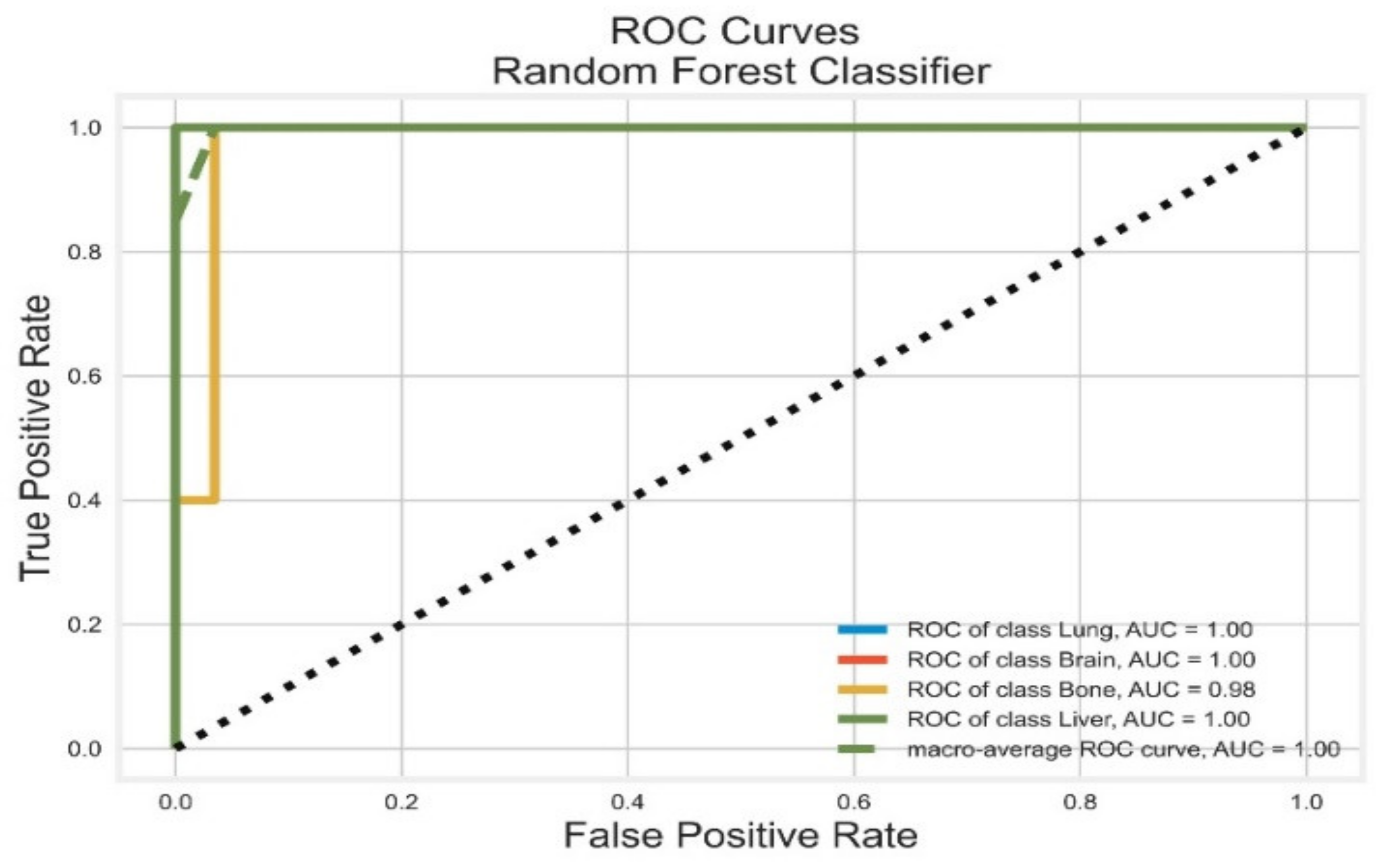
3.2. Random Forest Classifier
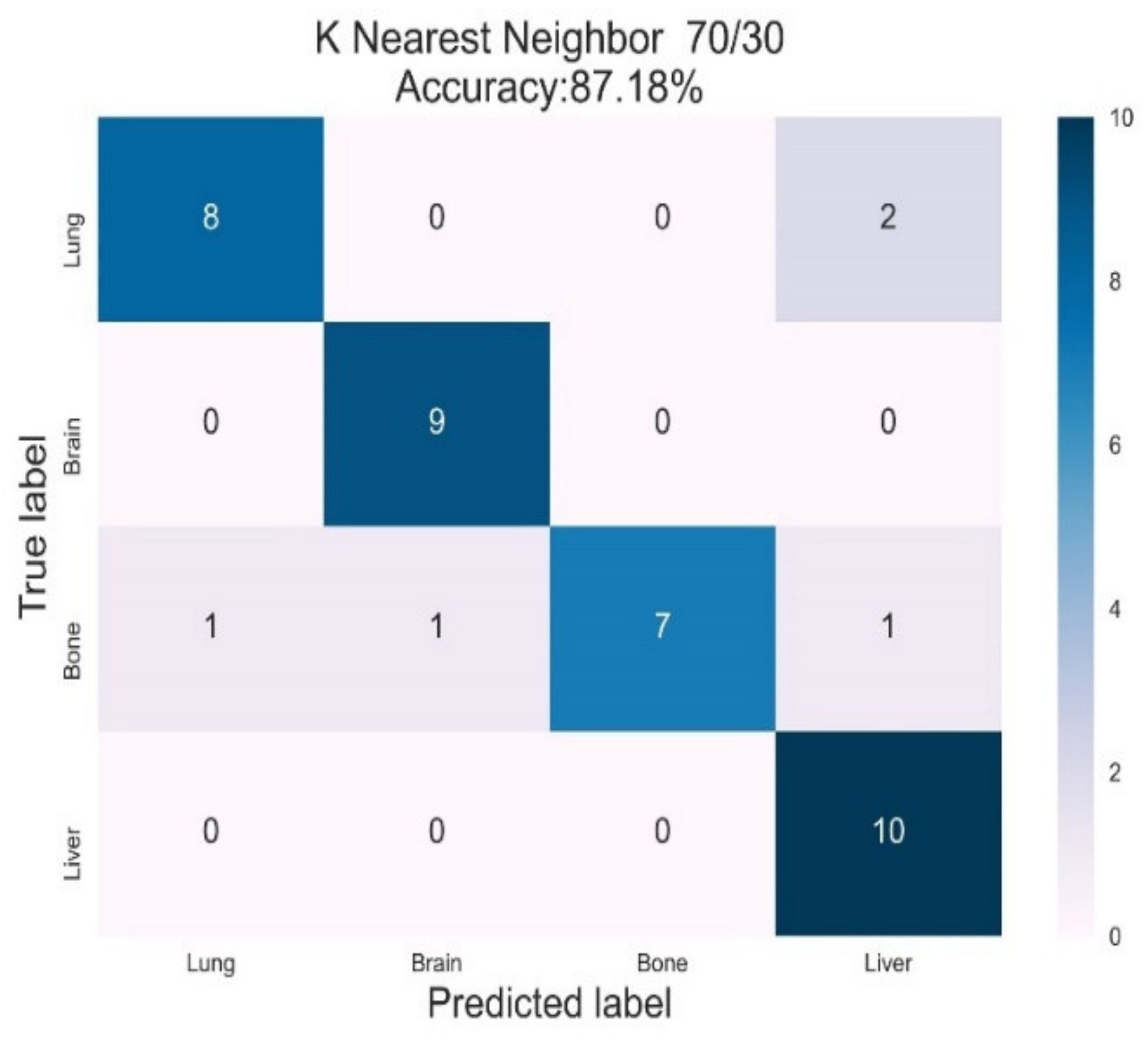
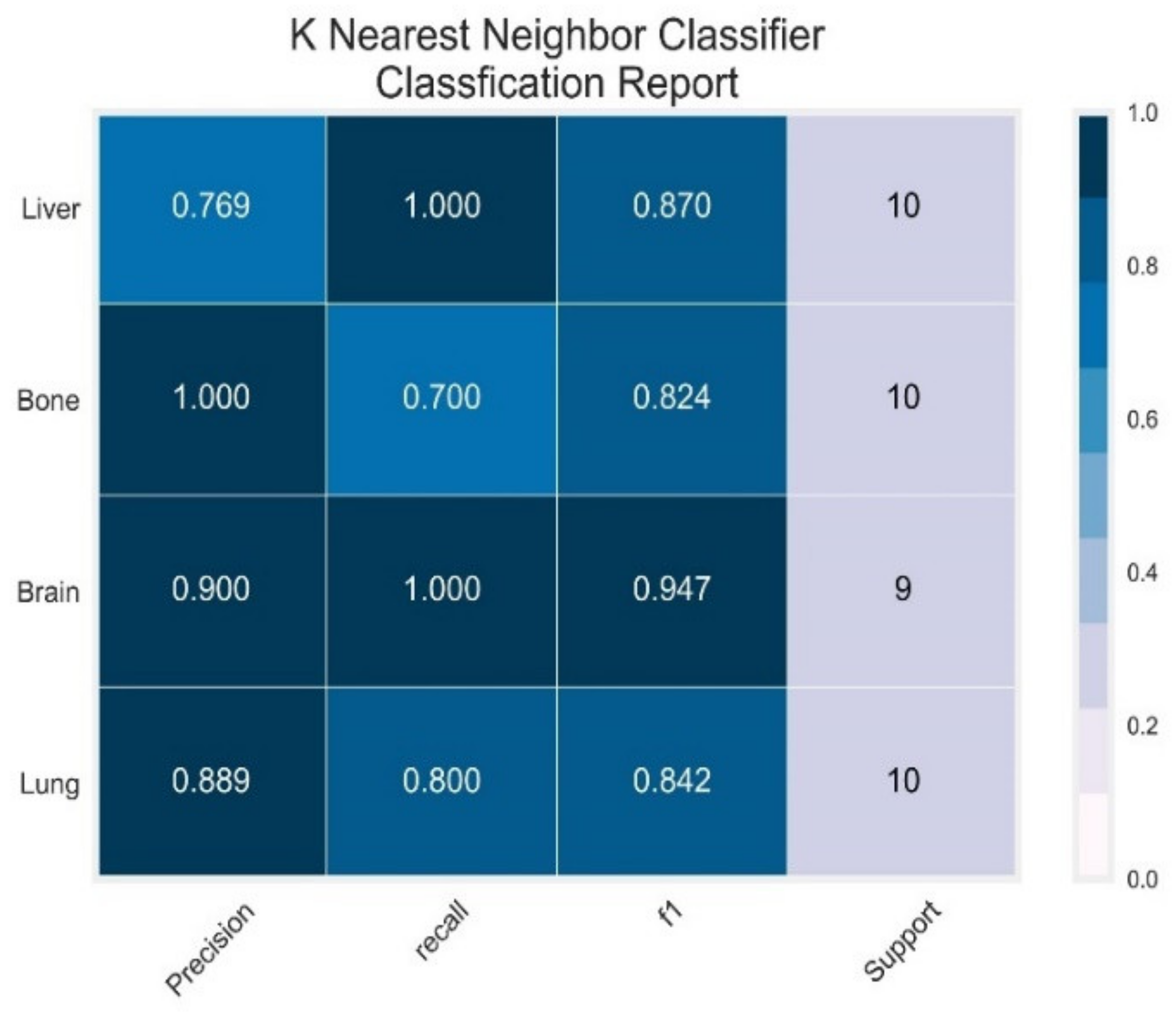
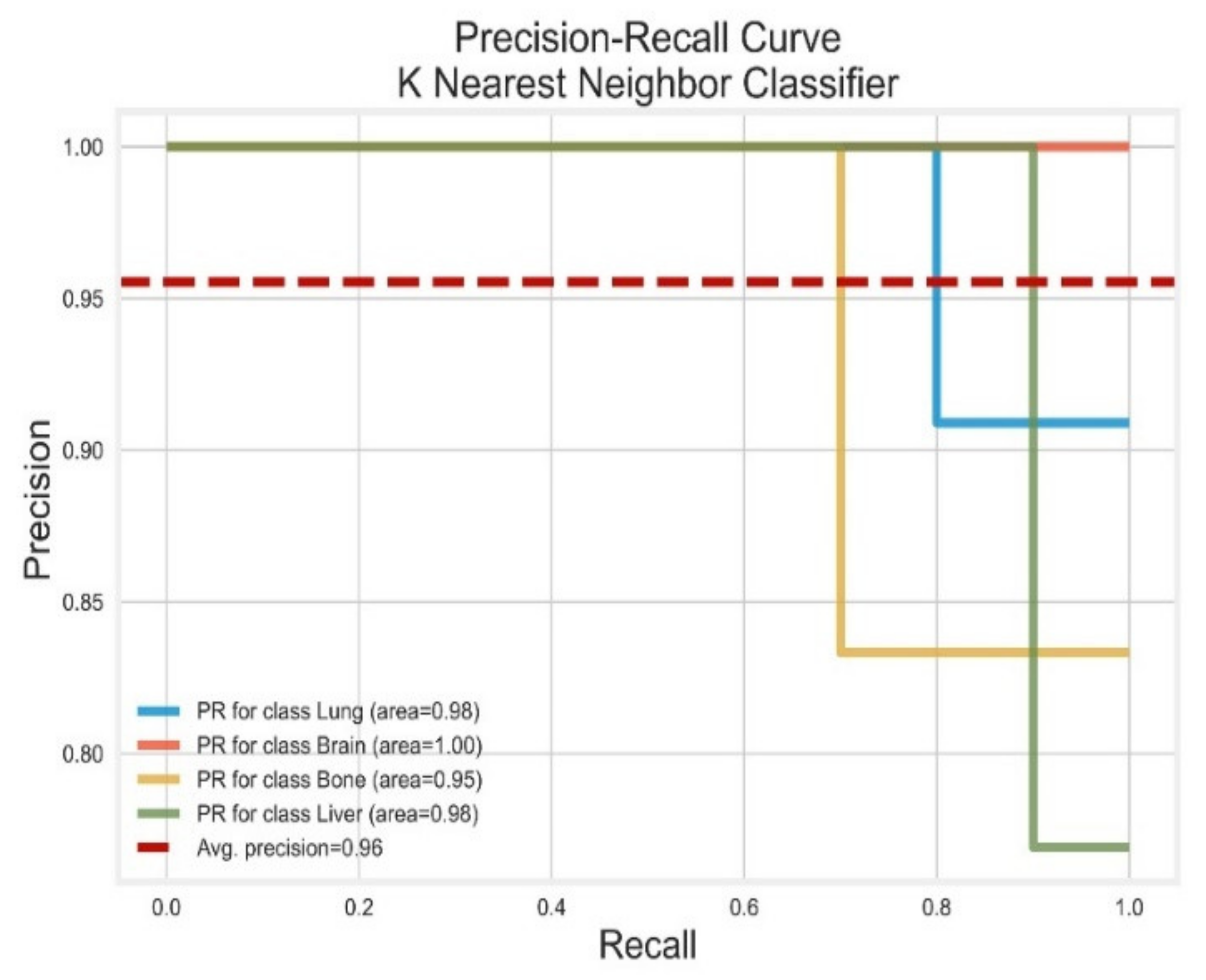
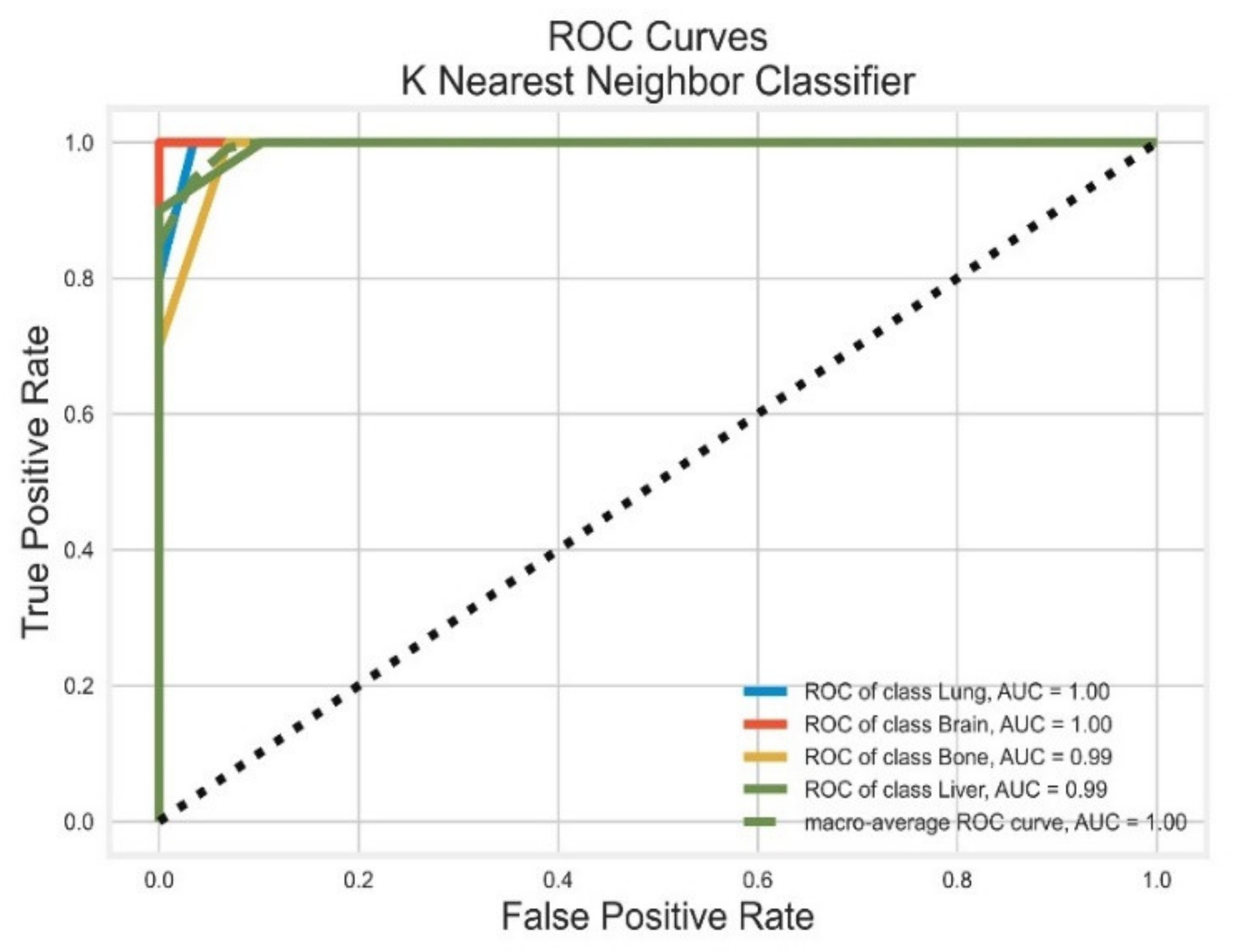
3.3. K-Nearest Neighbour Classifier
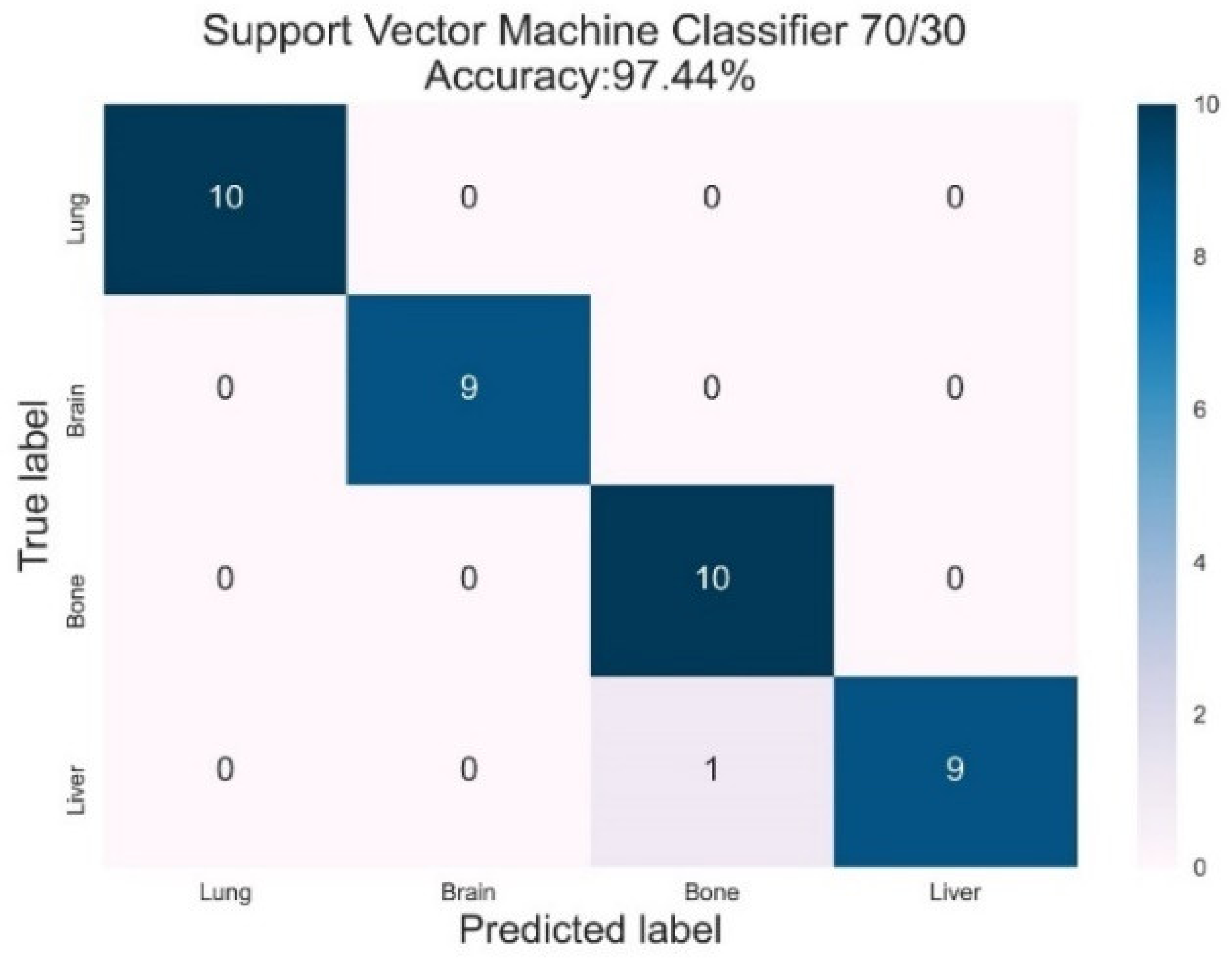
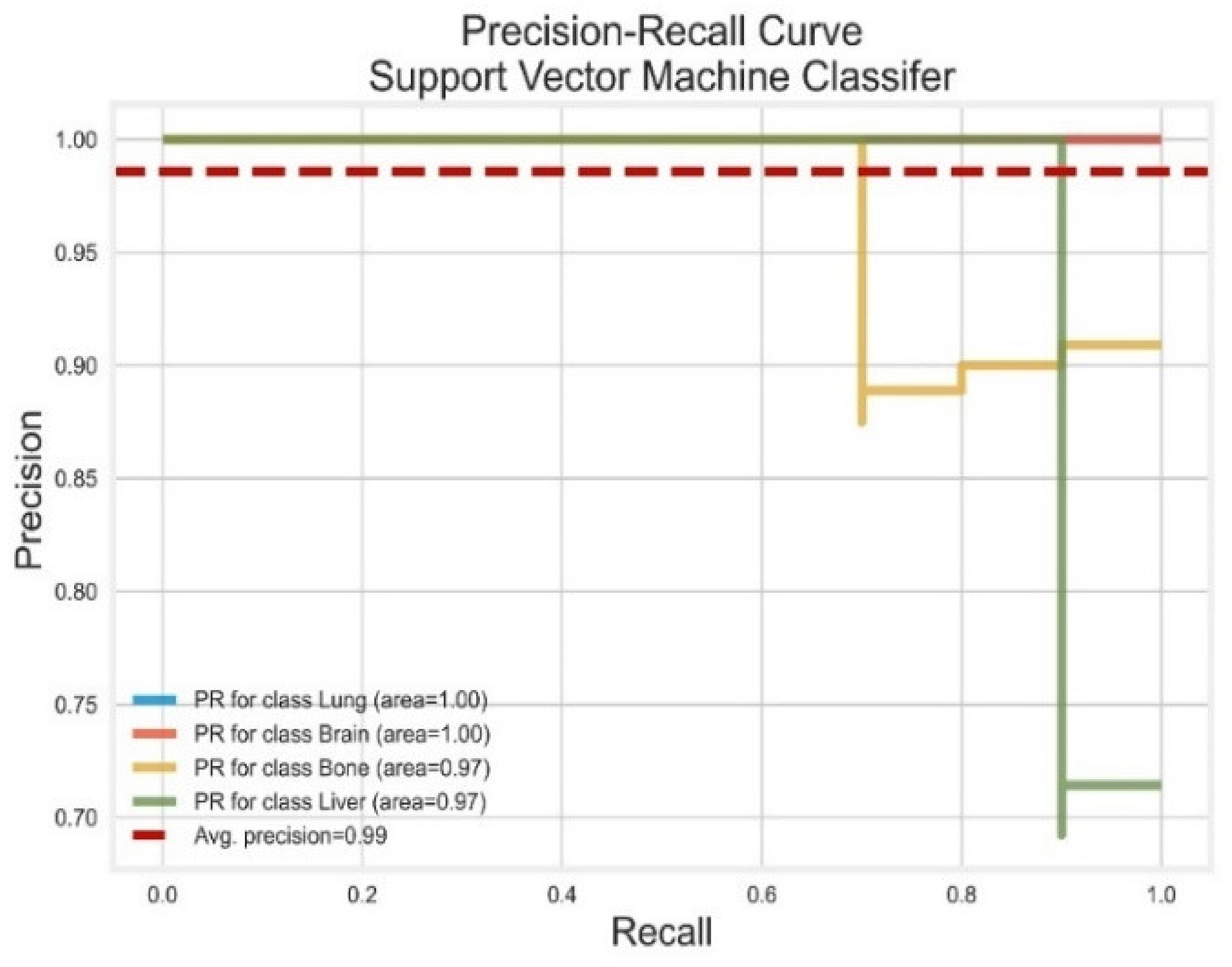
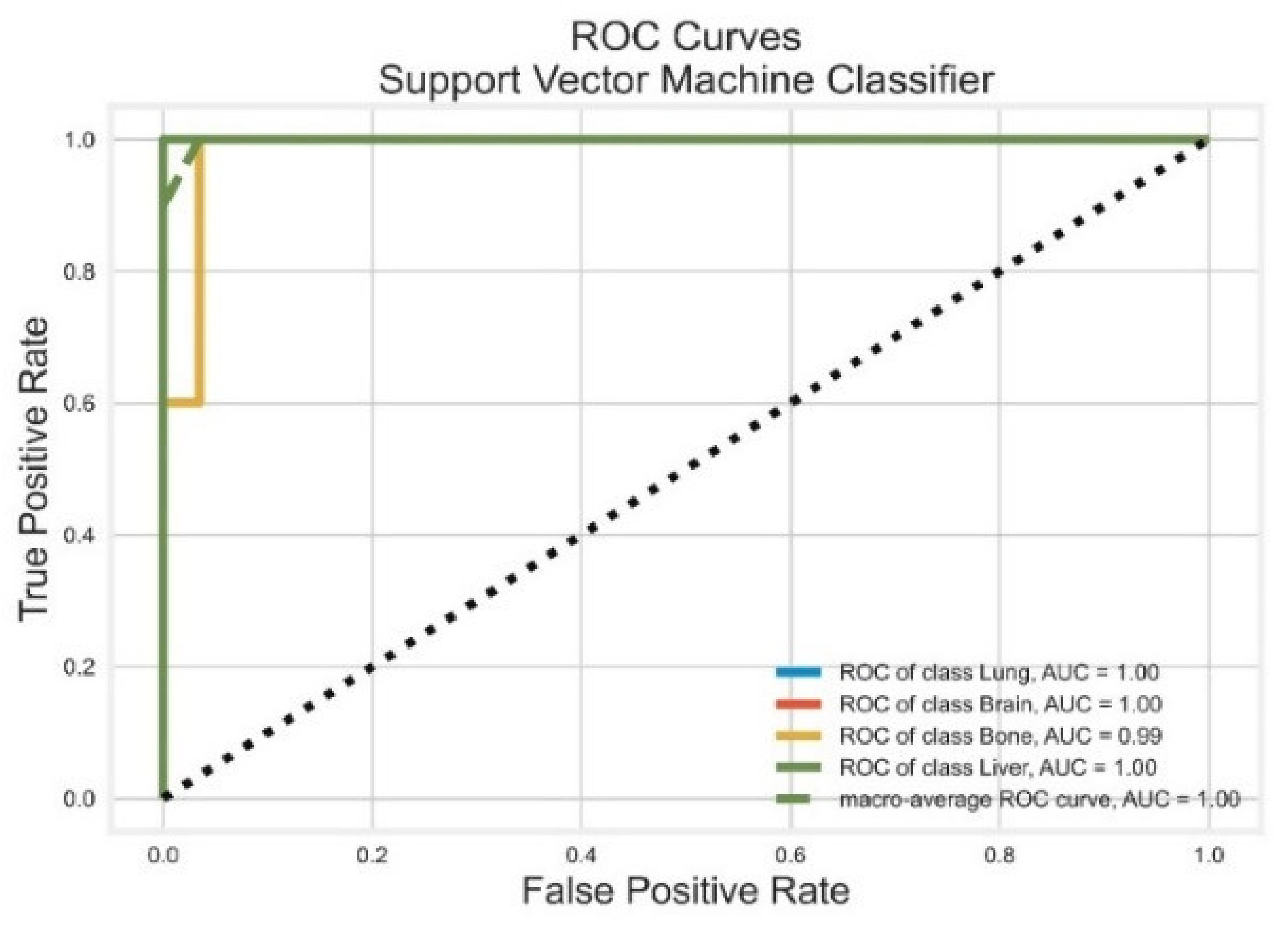
3.4. Support Vector Machines
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| BC | Breast Cancer |
| BCBoM | Breast Cancer Bone Metastasis |
| BCBrM | Breast Cancer Brain Metastasis |
| BCLiM | Breast Cancer Liver Metastasis |
| BCLuM | Breast Cancer Lung Metastasis |
| CTCs | Circulating Tumor cells |
| CV | Cross validation |
| DT | Decision Tree supervised learning algorithm |
| FN | False Negative |
| FP | False Positive |
| GEO | Gene expression omnibus |
| K-Means | Unsupervised learning algorithm that partition dataset into K number of clusters |
| KNN | K-Nearest Neighbors supervised learning algorithm |
| NCBI | National Center for Biotechnology Information |
| NRF | National Research Foundation |
| PR | Precision Recall |
| RF | Random Forrest supervised learning algorithm |
| RFECV | Recursive Feature Elimination with cross validation |
| ROC | Receiver operating characteristic curve |
| SMOTE | Synthetic object oversampling technique |
| SVM | Support Vector Machine supervised learning algorithm |
| TN | True Negative |
| TP | True Positive |
Appendix A. GSE14020
| ENTREZ_GENE_ID | Index | 1 | 10 | 100 | 1000 | 10,000 | 100,009,676 | … | 9990 | 9991 | 9992 | 9993 | 9994 | 9997 | Metastasis |
| 0 | GSM352095 | 8.2339 | 6.0683 | 7.8848 | 6.8721 | 6.6923 | 5.1057 | … | 5.2606 | 7.9440 | 6.4849 | 9.3538 | 6.5735 | 10.7023 | Lung |
| 1 | GSM352097 | 6.9053 | 6.8495 | 7.0798 | 9.1682 | 6.9491 | 5.3145 | … | 6.3746 | 7.6820 | 6.6459 | 8.7531 | 7.0091 | 8.8169 | Brain |
| 2 | GSM352098 | 7.7113 | 7.2329 | 6.5495 | 7.2243 | 6.2732 | 5.5823 | … | 5.9297 | 8.2616 | 6.5661 | 9.7786 | 7.6423 | 10.8695 | Brain |
| 3 | GSM352100 | 7.3770 | 6.7069 | 7.8392 | 7.3133 | 7.1324 | 5.1022 | … | 5.8855 | 7.6461 | 6.5335 | 8.9864 | 6.8501 | 9.6639 | Bone |
| 4 | GSM352101 | 7.4527 | 6.8992 | 6.4535 | 10.0303 | 6.4948 | 5.1645 | … | 5.7241 | 7.4800 | 6.7075 | 9.0687 | 7.0794 | 9.8207 | Brain |
| 5 | GSM352103 | 7.4528 | 6.8274 | 6.3866 | 10.8126 | 7.1971 | 5.4020 | … | 5.8136 | 7.3723 | 6.8072 | 8.3742 | 7.3350 | 9.8820 | Bone |
| 6 | GSM352105 | 7.8187 | 6.6937 | 9.4577 | 10.7817 | 6.6365 | 5.1032 | … | 6.1665 | 7.1127 | 6.6030 | 8.4056 | 7.4552 | 10.1645 | Bone |
| 7 | GSM352107 | 6.9245 | 6.4157 | 7.1858 | 7.8156 | 8.3712 | 5.0686 | … | 5.8752 | 7.5597 | 6.3730 | 8.8120 | 8.5782 | 10.5591 | Brain |
| 8 | GSM352109 | 7.3767 | 6.4297 | 7.1149 | 7.5414 | 6.6557 | 5.1151 | … | 5.7456 | 7.4064 | 6.3695 | 8.5874 | 6.9917 | 9.9465 | Bone |
| 9 | GSM352110 | 6.9355 | 6.8455 | 7.2018 | 8.0589 | 6.5079 | 5.2703 | … | 5.8681 | 9.3738 | 6.6825 | 9.0142 | 7.1229 | 9.2914 | Brain |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 55 | GSM352159 | NaN | 7.1089 | 6.6904 | 6.7784 | 5.1182 | NaN | … | 5.9607 | 7.0477 | 6.8578 | 7.4886 | 6.9233 | 8.5203 | Bone |
| 56 | GSM352160 | NaN | 6.5573 | 7.5134 | 6.8305 | 5.4619 | NaN | … | 6.1137 | 6.7245 | 6.8011 | 7.8224 | 6.6020 | 9.0305 | Lung |
| 57 | GSM352161 | NaN | 7.1461 | 6.9137 | 6.6266 | 5.0809 | NaN | … | 5.8923 | 6.5635 | 7.1701 | 8.3298 | 7.2257 | 9.5441 | Lung |
| 58 | GSM352162 | NaN | 7.6670 | 6.4741 | 7.0452 | 5.1741 | NaN | … | 5.7948 | 6.6691 | 6.7381 | 7.5607 | 7.8597 | 7.8423 | Liver |
| 59 | GSM352163 | NaN | 6.9949 | 7.4864 | 6.6844 | 5.5261 | NaN | … | 6.2890 | 6.7573 | 7.0070 | 7.5590 | 6.2727 | 9.0151 | Bone |
| 60 | GSM352164 | NaN | 6.5512 | 7.3306 | 7.4419 | 5.4275 | NaN | … | 5.8670 | 6.2873 | 6.7903 | 8.0515 | 7.1116 | 8.6275 | Lung |
| 61 | GSM352165 | NaN | 6.5440 | 7.3335 | 6.6873 | 5.9610 | NaN | … | 5.6563 | 6.9762 | 6.7983 | 8.1609 | 6.1113 | 9.2522 | Lung |
| 62 | GSM352166 | NaN | 6.9497 | 6.6913 | 6.6506 | 6.2395 | NaN | … | 5.9061 | 6.2398 | 7.0367 | 7.8028 | 6.8671 | 8.4663 | Lung |
| 63 | GSM352167 | NaN | 6.7676 | 7.1345 | 7.6319 | 6.5522 | NaN | … | 5.9233 | 6.5831 | 6.9459 | 8.0467 | 6.3893 | 8.5806 | Bone |
| 64 | GSM352168 | NaN | 7.4883 | 6.9053 | 6.8512 | 5.6716 | NaN | … | 6.5621 | 6.0487 | 7.0821 | 7.7147 | 6.9370 | 9.2354 | Lung |
| 65 rows × 20,488 columns. | |||||||||||||||
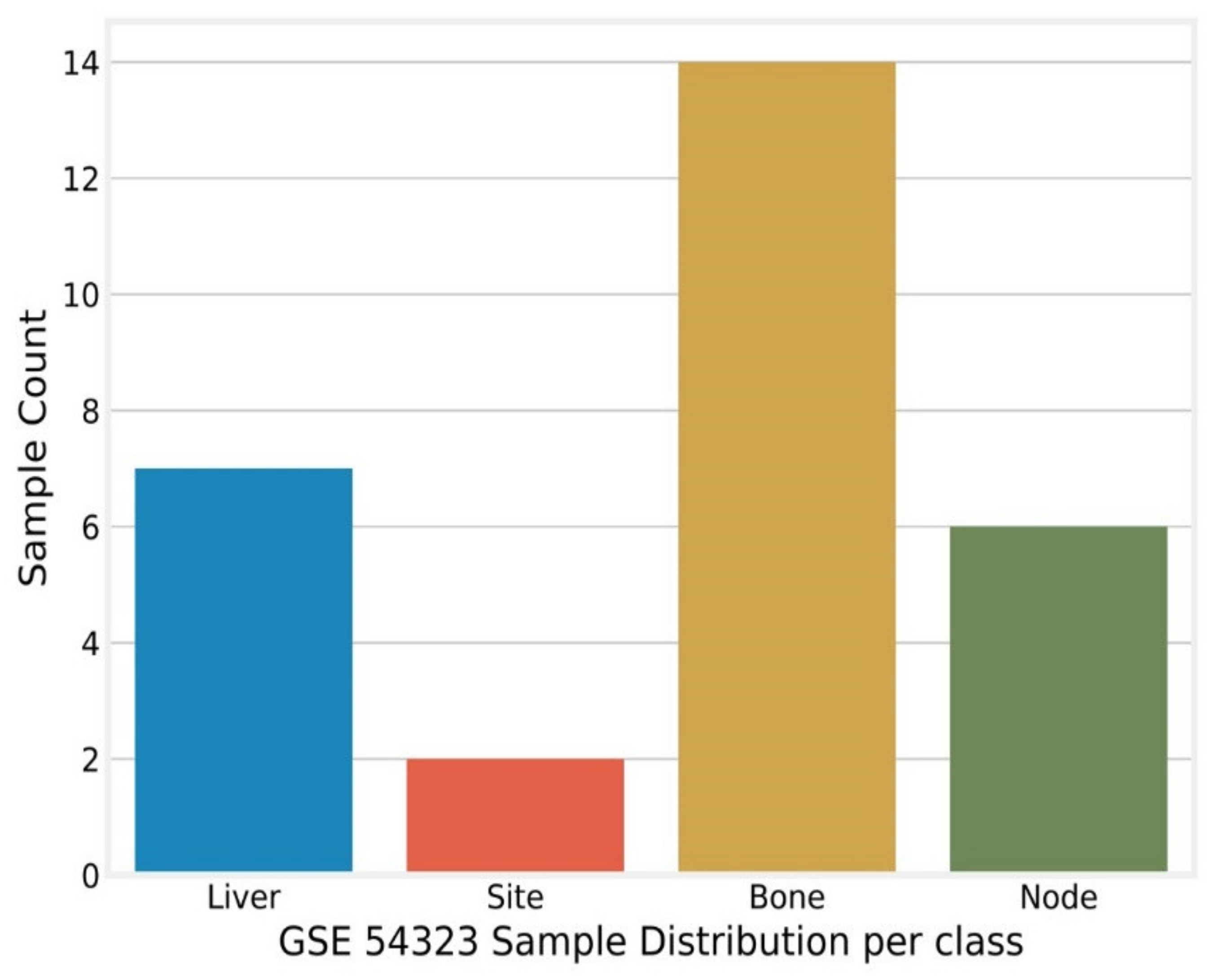
Appendix B. GSE54323
| ENTREZ_GENE_ID | Index | 1 | 10 | 100 | 1000 | 10,000 | 100,009,676 | … | 9990 | 9991 | 9992 | 9993 | 9994 | 9997 | Metastasis |
| 0 | GSM1312928 | 7.5602 | 6.4469 | 7.3371 | 5.7804 | 4.4252 | 4.9310 | … | 6.4865 | 8.2751 | 5.9836 | 8.2346 | 6.8023 | 9.9452 | Liver |
| 1 | GSM1312929 | 10.3885 | 8.2872 | 7.8423 | 7.3761 | 4.6025 | 5.1180 | … | 6.0557 | 6.9495 | 6.7754 | 8.0115 | 5.8184 | 9.1237 | Liver |
| 2 | GSM1312930 | 6.7133 | 5.8366 | 5.7955 | 6.9065 | 4.6457 | 4.8781 | … | 5.4504 | 8.9469 | 6.3186 | 8.3371 | 6.3372 | 9.3204 | Site |
| 3 | GSM1312931 | 6.2121 | 6.3439 | 5.7062 | 5.9918 | 4.3514 | 4.8626 | … | 6.3488 | 9.0194 | 6.4219 | 8.1964 | 6.2462 | 10.2259 | Site |
| 4 | GSM1312932 | 5.8098 | 4.9986 | 6.9900 | 5.3402 | 4.6099 | 4.9493 | … | 5.2374 | 7.8907 | 6.1864 | 8.0367 | 6.0154 | 10.5537 | Bone |
| 5 | GSM1312933 | 5.6922 | 5.0370 | 6.0000 | 5.3512 | 4.2550 | 4.9094 | … | 6.7374 | 8.4135 | 6.1085 | 8.2670 | 5.8228 | 9.6436 | Bone |
| 6 | GSM1312934 | 6.2121 | 5.4978 | 7.2723 | 5.3844 | 4.3429 | 5.7149 | … | 5.9428 | 9.2940 | 6.2257 | 8.3165 | 6.6325 | 9.7894 | Node |
| 7 | GSM1312935 | 6.4032 | 4.8821 | 7.3576 | 5.5975 | 5.3479 | 4.9806 | … | 6.1142 | 7.3487 | 6.1657 | 8.0086 | 6.4750 | 9.3352 | Node |
| 8 | GSM1312936 | 6.5354 | 5.3353 | 6.2503 | 5.4279 | 4.3448 | 5.2540 | … | 5.5127 | 8.1144 | 6.2992 | 8.4049 | 6.3864 | 9.6644 | Node |
| 9 | GSM1312937 | 6.1965 | 5.0868 | 7.0903 | 5.2549 | 4.9637 | 4.9261 | … | 6.8642 | 7.6783 | 7.0730 | 8.4742 | 5.4469 | 10.6389 | Node |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 19 | GSM1312947 | 6.0102 | 4.8673 | 7.4504 | 5.0322 | 5.0102 | 4.9572 | … | 8.3811 | 8.8105 | 6.2732 | 8.1065 | 6.2530 | 10.7216 | Bone |
| 20 | GSM1312948 | 6.0744 | 5.0904 | 6.7679 | 5.7245 | 4.4632 | 4.7979 | … | 6.8297 | 8.1070 | 6.5280 | 8.1100 | 5.9623 | 8.9717 | Bone |
| 21 | GSM1312949 | 6.7086 | 4.8407 | 6.6089 | 5.5950 | 4.3482 | 5.0114 | … | 6.9696 | 7.1899 | 6.3186 | 8.2858 | 5.6671 | 8.6960 | Bone |
| 22 | GSM1312950 | 5.7694 | 5.5062 | 7.2207 | 5.2119 | 5.0936 | 5.3236 | … | 5.5929 | 8.3197 | 6.0860 | 8.1592 | 6.9327 | 9.4827 | Node |
| 23 | GSM1312951 | 6.1343 | 5.4438 | 6.6683 | 5.5099 | 6.1355 | 5.5816 | … | 5.9490 | 8.3783 | 6.4996 | 8.3447 | 6.4786 | 9.3974 | Node |
| 24 | GSM1312952 | 5.8563 | 4.8592 | 5.6757 | 5.3235 | 4.4993 | 5.2810 | … | 5.6794 | 8.8107 | 6.2094 | 8.1863 | 5.8603 | 8.3529 | Liver |
| 25 | GSM1312953 | 8.8499 | 6.0498 | 5.9915 | 6.1030 | 4.3604 | 5.0026 | … | 5.3545 | 8.1970 | 6.2779 | 8.2310 | 5.9560 | 9.5760 | Bone |
| 26 | GSM1312954 | 6.3137 | 4.7653 | 5.1842 | 5.2177 | 4.2325 | 5.7308 | … | 5.2766 | 9.3691 | 6.1371 | 8.0164 | 6.1084 | 8.2146 | Bone |
| 27 | GSM1312955 | 5.9951 | 5.0517 | 6.4163 | 5.2682 | 4.3745 | 4.8983 | … | 6.2287 | 8.7048 | 6.0669 | 8.4942 | 5.6814 | 9.5736 | Bone |
| 28 | GSM1312956 | 6.0545 | 4.7172 | 6.9508 | 5.1912 | 4.3765 | 4.8779 | … | 7.6965 | 8.8376 | 6.4431 | 8.2028 | 6.5684 | 9.2403 | Bone |
| 29 rows × 20,488 columns. | |||||||||||||||
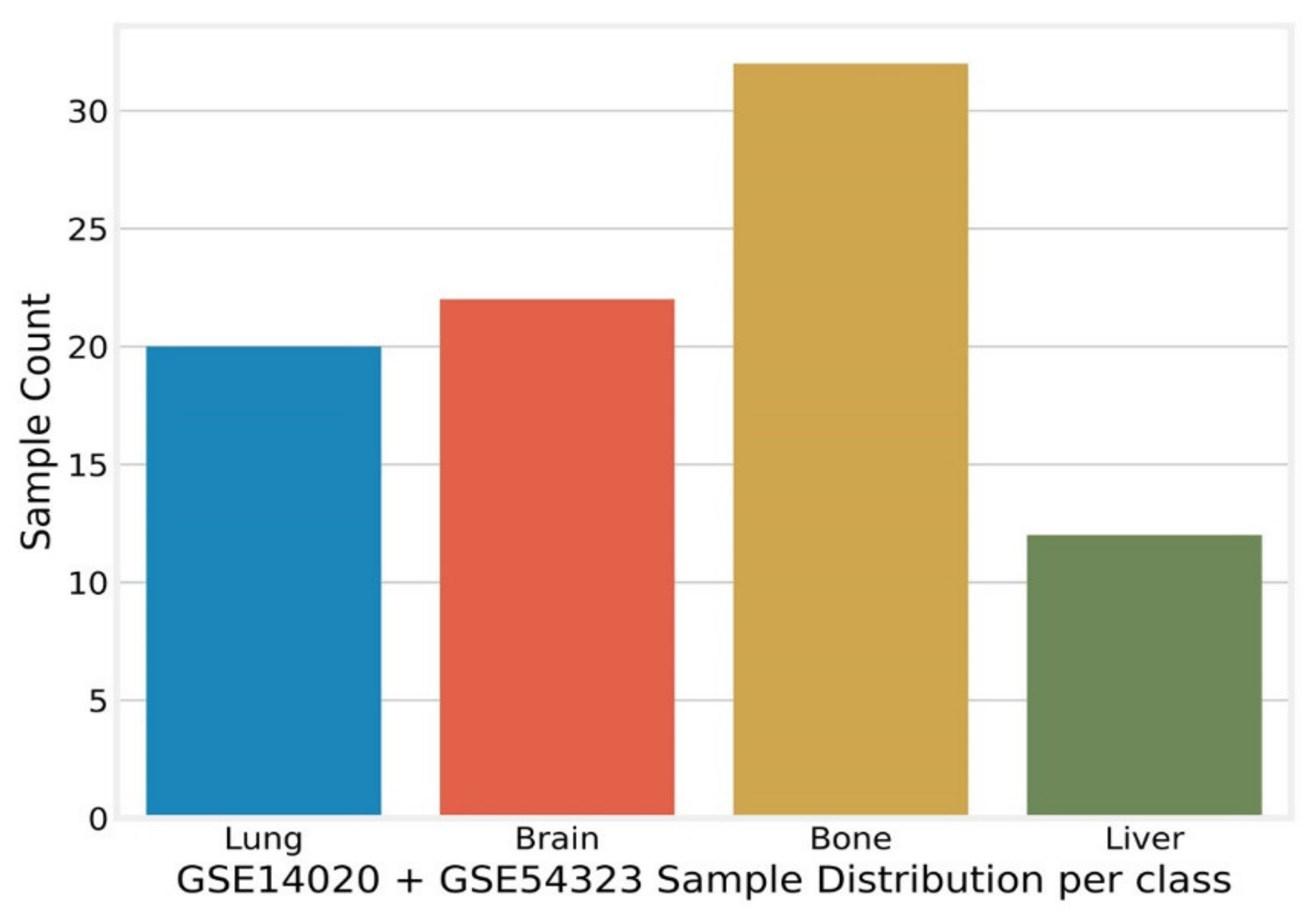
Appendix C. Merged and Normalized Dataset
| 1 | 10 | 100 | 1000 | 10000 | 1 × 108 | 10001 | ... | 999 | 9990 | 9991 | 9992 | 9993 | 9994 | 9997 | |
| 0 | 1.1073 | −0.5584 | 1.4254 | −0.1679 | 0.968 | −0.7103 | 0.3433 | ... | 0.235 | −1.4055 | 0.6487 | −0.7894 | 1.7514 | −0.47 | 1.4987 |
| 1 | −0.4006 | 0.3714 | 0.2092 | 1.7604 | 1.2383 | 0.2766 | 0.7643 | ... | 0.5823 | 0.5341 | 0.3564 | −0.2172 | 0.7292 | 0.1851 | −0.5383 |
| 2 | 0.5142 | 0.8277 | −0.5921 | 0.1279 | 0.527 | 1.5429 | 0.784 | ... | 1.3263 | −0.2405 | 1.0032 | −0.5009 | 2.4741 | 1.1371 | 1.6794 |
| 3 | 0.1348 | 0.2017 | 1.3565 | 0.2027 | 1.4312 | −0.7269 | 1.0486 | ... | 0.8783 | −0.3174 | 0.3164 | −0.6167 | 1.1261 | −0.0541 | 0.3768 |
| 4 | 0.2206 | 0.4306 | −0.7371 | 2.4844 | 0.7602 | −0.4321 | 1.08 | ... | 0.8878 | −0.5984 | 0.131 | 0.002 | 1.2663 | 0.2907 | 0.5462 |
| 5 | 0.2208 | 0.3451 | −0.8381 | 3.1413 | 1.4992 | 0.6906 | 1.4515 | ... | −3.5736 | −0.4426 | 0.0108 | 0.3564 | 0.0845 | 0.6751 | 0.6125 |
| 6 | 0.6361 | 0.186 | 3.8018 | 3.1154 | 0.9093 | −0.7219 | 2.3365 | ... | −3.1912 | 0.1717 | −0.2788 | −0.3698 | 0.138 | 0.8558 | 0.9176 |
| 7 | −0.3789 | −0.145 | 0.3693 | 0.6245 | 2.7348 | −0.8856 | 1.0825 | ... | 1.0557 | −0.3354 | 0.2199 | −1.1873 | 0.8295 | 2.5444 | 1.3439 |
| 8 | 0.1344 | −0.1282 | 0.2622 | 0.3942 | 0.9295 | −0.666 | 1.3149 | ... | 0.9476 | −0.561 | 0.0489 | −1.1998 | 0.4473 | 0.1588 | 0.6821 |
| 9 | −0.3664 | 0.3666 | 0.3935 | 0.8288 | 0.774 | 0.0678 | 0.6543 | ... | 2.0178 | −0.3477 | 2.2444 | −0.087 | 1.1735 | 0.3561 | −0.0257 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 76 | −1.6765 | −2.1117 | 0.5075 | −1.1132 | −0.8302 | −2.4271 | −1.1618 | ... | −0.8085 | 2.8795 | −0.3754 | −1.3423 | −0.3784 | −1.3842 | 1.1868 |
| 77 | −1.7519 | −1.7871 | −0.2358 | −1.4794 | −0.1956 | −0.0349 | −1.4196 | ... | −0.6919 | 2.6152 | 1.5135 | −1.5502 | −0.057 | −0.3818 | 0.9789 |
| 78 | −1.4165 | −1.9877 | 0.7691 | −1.713 | −0.8021 | −1.4121 | −0.6681 | ... | −1.1026 | 4.0275 | 1.6157 | −1.5423 | −0.371 | −0.952 | 1.5195 |
| 79 | −1.3437 | −1.7222 | −0.2621 | −1.1316 | −1.3777 | −2.1654 | −1.7002 | ... | −0.9386 | 1.3265 | 0.8307 | −0.6363 | −0.365 | −1.389 | −0.3711 |
| 80 | −0.6238 | −2.0195 | −0.5024 | −1.2403 | −1.4987 | −1.1558 | −1.9665 | ... | −0.7943 | 1.5701 | −0.1927 | −1.3807 | −0.0658 | −1.8329 | −0.6689 |
| 81 | −1.5912 | −1.9974 | −1.9121 | −1.4683 | −1.3397 | 0.1184 | −1.0834 | ... | 0.0965 | −0.6762 | 1.616 | −1.7688 | −0.2352 | −1.5423 | −1.0397 |
| 82 | 1.8065 | −0.5804 | −1.4351 | −0.8137 | −1.4859 | −1.1977 | −1.0668 | ... | 0.8924 | −1.2419 | 0.9312 | −1.5253 | −0.1591 | −1.3984 | 0.2818 |
| 83 | −1.0721 | −2.1091 | −2.6548 | −1.5571 | −1.6205 | 2.2447 | −0.0992 | ... | 0.7633 | −1.3775 | 2.2391 | −2.0259 | −0.5243 | −1.1693 | −1.1891 |
| 84 | −1.4337 | −1.7683 | −0.7933 | −1.5147 | −1.4711 | −1.6908 | −1.2391 | ... | 0.1758 | 0.2801 | 1.4978 | −2.2756 | 0.2887 | −1.8114 | 0.2792 |
| 85 | −1.3663 | −2.1664 | 0.0143 | −1.5794 | −1.4689 | −1.7872 | −1.4259 | ... | −1.3731 | 2.8356 | 1.646 | −0.938 | −0.207 | −0.4777 | −0.0809 |
| 86 rows × 20,486 columns. | |||||||||||||||
Appendix D. Dataset after Removing Correlated Features
| 1 | 10 | 100 | 1000 | 10,000 | 1 × 108 | 10,001 | … | 9961 | 9962 | 997 | 9973 | 9984 | 999 | 9997 | |
| 0 | 1.1073 | −0.5584 | 1.4254 | −0.1679 | 0.968 | −0.7103 | 0.3433 | … | 0.2706 | 0.4551 | 1.6566 | 1.1436 | −0.4051 | 0.235 | 1.4987 |
| 1 | −0.4006 | 0.3714 | 0.2092 | 1.7604 | 1.2383 | 0.2766 | 0.7643 | … | 0.4908 | 0.4133 | 1.3221 | 0.6164 | −0.2804 | 0.5823 | −0.5383 |
| 2 | 0.5142 | 0.8277 | −0.5921 | 0.1279 | 0.527 | 1.5429 | 0.784 | … | 1.1641 | 0.4141 | 0.6974 | 0.9285 | 0.2504 | 1.3263 | 1.6794 |
| 3 | 0.1348 | 0.2017 | 1.3565 | 0.2027 | 1.4312 | −0.7269 | 1.0486 | … | 1.7457 | −0.3479 | 0.0111 | 0.5555 | −1.3193 | 0.8783 | 0.3768 |
| 4 | 0.2206 | 0.4306 | −0.7371 | 2.4844 | 0.7602 | −0.4321 | 1.08 | … | 1.4034 | −0.6757 | 0.8695 | 0.2926 | 3.6107 | 0.8878 | 0.5462 |
| 5 | 0.2208 | 0.3451 | −0.8381 | 3.1413 | 1.4992 | 0.6906 | 1.4515 | … | 0.1724 | 0.5311 | −0.276 | 0.4835 | 0.33 | −3.5736 | 0.6125 |
| 6 | 0.6361 | 0.186 | 3.8018 | 3.1154 | 0.9093 | −0.7219 | 2.3365 | … | 0.3219 | 0.6182 | −0.4591 | 0.0423 | −0.6683 | −3.1912 | 0.9176 |
| 7 | −0.3789 | −0.145 | 0.3693 | 0.6245 | 2.7348 | −0.8856 | 1.0825 | … | 0.0195 | −0.1824 | −0.3309 | 2.046 | −0.0084 | 1.0557 | 1.3439 |
| 8 | 0.1344 | −0.1282 | 0.2622 | 0.3942 | 0.9295 | −0.666 | 1.3149 | … | 0.9737 | −0.5723 | −0.6066 | 0.7488 | −0.1313 | 0.9476 | 0.6821 |
| 9 | −0.3664 | 0.3666 | 0.3935 | 0.8288 | 0.774 | 0.0678 | 0.6543 | … | 0.3707 | 0.221 | 0.2925 | 0.1104 | −0.2957 | 2.0178 | −0.0257 |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 76 | −1.6765 | −2.1117 | 0.5075 | −1.1132 | −0.8302 | −2.4271 | −1.1618 | … | −0.1819 | −1.9179 | 2.44 | 0.735 | −0.7166 | −0.8085 | 1.1868 |
| 77 | −1.7519 | −1.7871 | −0.2358 | −1.4794 | −0.1956 | −0.0349 | −1.4196 | … | 0.0729 | −0.4649 | −2.2167 | −2.4931 | 1.0378 | −0.6919 | 0.9789 |
| 78 | −1.4165 | −1.9877 | 0.7691 | −1.713 | −0.8021 | −1.4121 | −0.6681 | … | 0.9578 | −0.3947 | −1.3783 | −1.6917 | 0.2525 | −1.1026 | 1.5195 |
| 79 | −1.3437 | −1.7222 | −0.2621 | −1.1316 | −1.3777 | −2.1654 | −1.7002 | … | 0.1721 | −1.5257 | 1.3317 | −1.0251 | 0.5284 | −0.9386 | −0.3711 |
| 80 | −0.6238 | −2.0195 | −0.5024 | −1.2403 | −1.4987 | −1.1558 | −1.9665 | … | 0.5117 | −1.6121 | 2.2701 | −0.6082 | 0.2498 | −0.7943 | −0.6689 |
| 81 | −1.5912 | −1.9974 | −1.9121 | −1.4683 | −1.3397 | 0.1184 | −1.0834 | … | −0.0482 | −1.9324 | −0.947 | −1.6412 | 0.3456 | 0.0965 | −1.0397 |
| 82 | 1.8065 | −0.5804 | −1.4351 | −0.8137 | −1.4859 | −1.1977 | −1.0668 | … | 0.2392 | 0.4666 | −0.4142 | 2.4051 | 0.4809 | 0.8924 | 0.2818 |
| 83 | −1.0721 | −2.1091 | −2.6548 | −1.5571 | −1.6205 | 2.2447 | −0.0992 | … | −0.4106 | −1.118 | −2.1279 | −1.5875 | 0.4401 | 0.7633 | −1.1891 |
| 84 | −1.4337 | −1.7683 | −0.7933 | −1.5147 | −1.4711 | −1.6908 | −1.2391 | … | −1.3745 | −2.1706 | −0.6876 | −0.8633 | 1.3487 | 0.1758 | 0.2792 |
| 85 | −1.3663 | −2.1664 | 0.0143 | −1.5794 | −1.4689 | −1.7872 | −1.4259 | … | 0.2409 | −2.3795 | 0.45 | −0.2579 | 0.5958 | −1.3731 | −0.0809 |
| 86 rows × 6602 columns. | |||||||||||||||
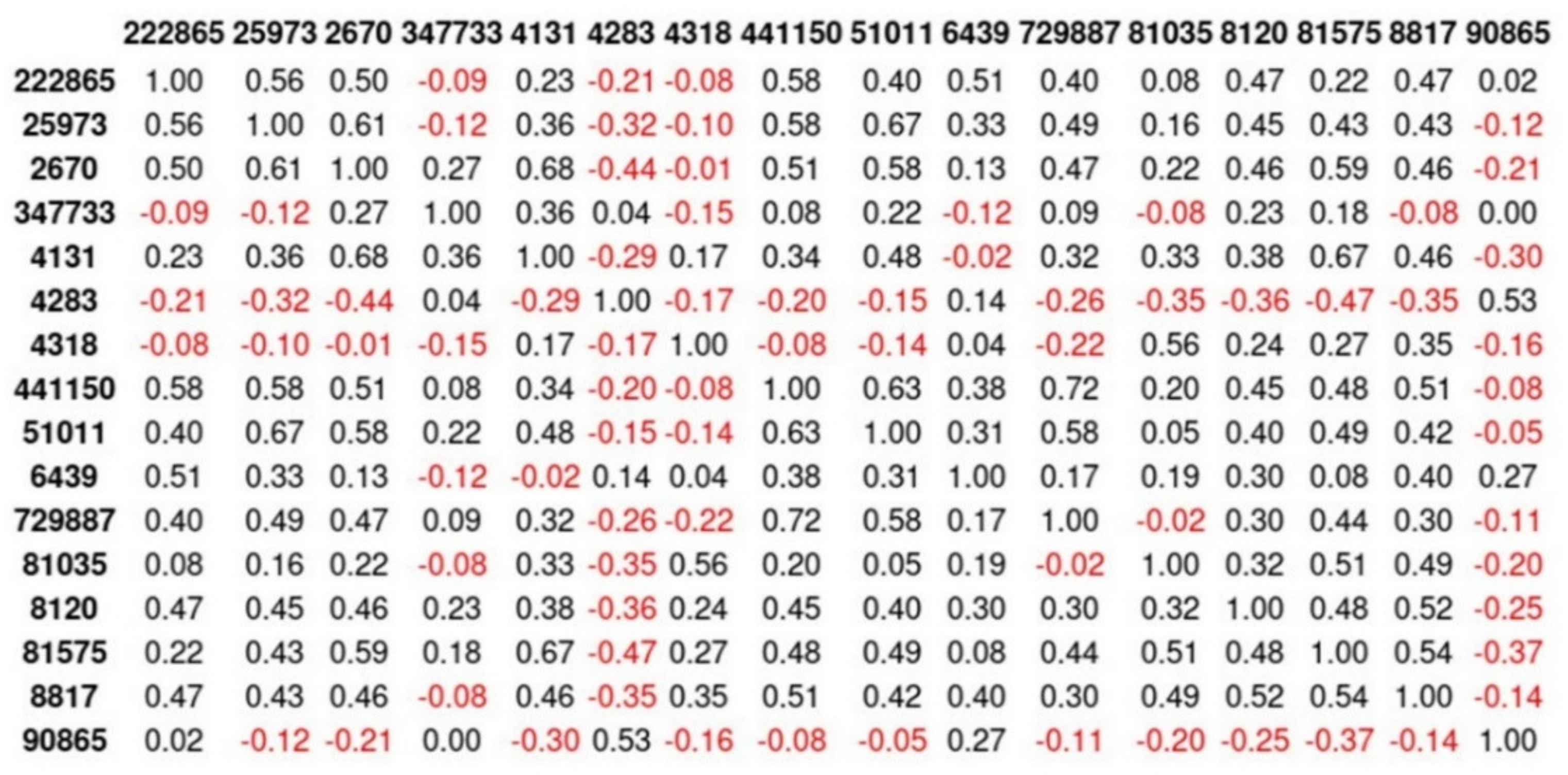
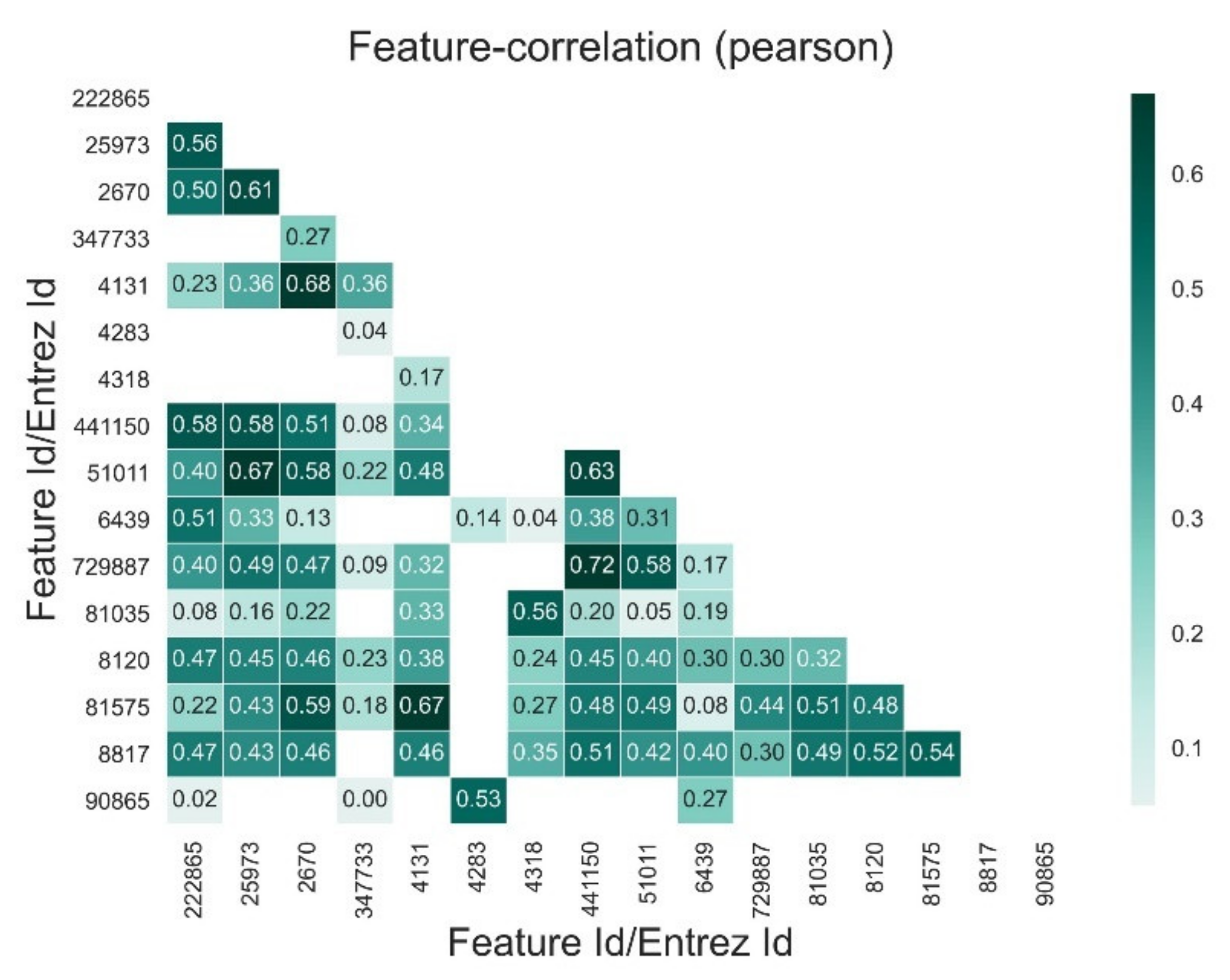
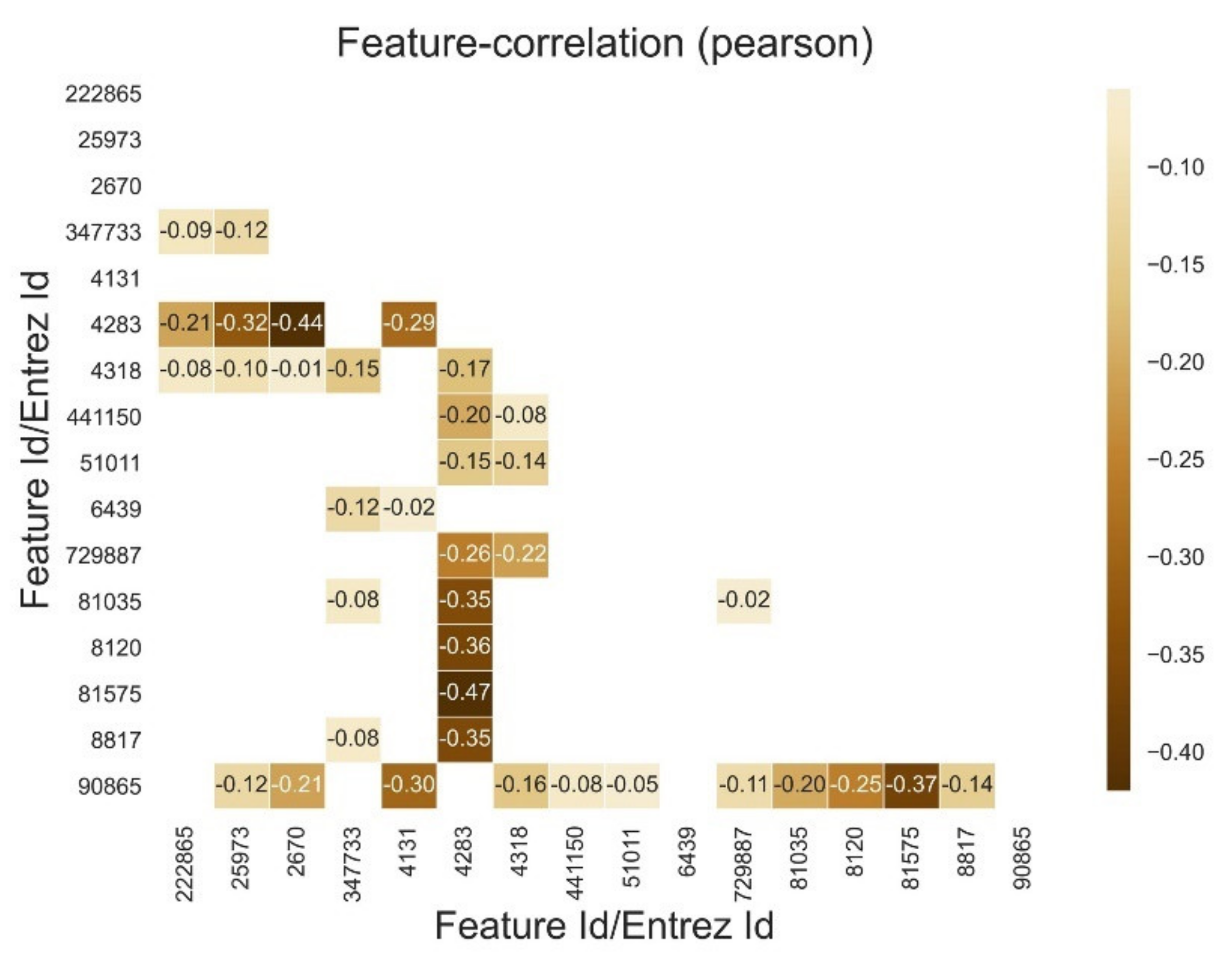
Appendix E. Reduced Dimensions after RFECV
| 222,865 | 25,973 | 2670 | 347,733 | 4131 | 4283 | 4318 | 441,150 | 51,011 | 6439 | 729,887 | 81,035 | 8120 | 81,575 | 8817 | 90,865 | |
| 0 | 0.198 | 0.9758 | 0.3665 | −0.2696 | 1.0458 | 0.9783 | −0.6444 | 3.782 | 3.171 | 0.3832 | 4.6323 | −0.4311 | −0.2163 | −0.1137 | −0.1295 | 0.7515 |
| 1 | −0.0865 | 1.1 | 0.7864 | 3.312 | 1.2416 | 0.5305 | 0.9294 | 0.6143 | 0.9198 | 0.1937 | −0.1079 | −0.3811 | 4.3088 | 1.2101 | −0.2767 | −0.4483 |
| 2 | 1.9646 | 1.1233 | 1.0511 | −0.0854 | 0.9364 | −0.79 | −1.2065 | 0.9356 | 0.1308 | −0.0736 | 2.1 | −0.71 | 0.2795 | 0.9134 | −0.3304 | −0.142 |
| 3 | −1.192 | −0.6367 | 0.2691 | −0.4562 | 1.7566 | −0.7846 | 1.4329 | −0.7015 | 0.4727 | −0.1349 | −0.7771 | 1.5955 | −0.3288 | 2.6731 | 3.8883 | 1.3303 |
| 4 | 0.1266 | −0.0604 | 1.7196 | 0.0398 | 1.8735 | −0.659 | 0.0221 | 0.1035 | 0.4401 | −0.0272 | 2.0996 | −0.2556 | −0.4302 | −0.0581 | 0.8987 | −0.5004 |
| 5 | −0.5906 | 0.3491 | 1.4248 | 0.2006 | 2.0667 | −0.7052 | −0.5249 | −0.2215 | −0.3393 | 0.1466 | −0.8944 | 0.6907 | −0.2445 | −0.2771 | −0.0903 | −0.4262 |
| 6 | −0.8871 | 0.1041 | 0.1751 | −0.7043 | 1.6466 | −0.3017 | 0.1538 | −0.4897 | 0.4002 | 0.0265 | −0.4333 | 0.4794 | −0.0686 | 1.0238 | −0.2101 | −0.2477 |
| 7 | 0.4481 | 0.0621 | 0.9934 | 1.1176 | 1.0911 | −0.7203 | 0.0915 | 1.7954 | 1.4472 | −0.0197 | 2.5998 | −0.3057 | −0.5338 | 1.8907 | 0.3507 | −0.4034 |
| 8 | −0.2063 | 1.0118 | 0.0792 | −0.7138 | 1.0369 | 1.4341 | 1.8265 | −0.2083 | 0.2059 | −0.0171 | −0.8552 | 1.9019 | −0.5076 | 0.847 | 0.2876 | 0.1463 |
| 9 | −0.2731 | 1.155 | 2.0704 | 1.0202 | 2.5062 | −0.6621 | −0.5338 | −0.1218 | 0.7685 | 0.1553 | 0.1663 | −0.5509 | −0.2045 | 1.5289 | 0.1918 | −0.2485 |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 76 | −1.012 | −1.9397 | −1.1871 | −0.9757 | −1.8074 | −0.5485 | 1.1886 | −1.6693 | −1.1848 | −0.9253 | −0.8543 | −0.6911 | −0.4686 | −0.2061 | −1.3553 | −0.7851 |
| 77 | −1.9576 | −0.0641 | −1.302 | −0.7803 | 0.1777 | 0.6541 | 1.6583 | −1.4042 | −0.9199 | −0.9623 | −1.659 | 0.5504 | −0.8571 | −0.2125 | −1.4715 | −1.1276 |
| 78 | −1.786 | −1.2262 | −1.3476 | −0.3278 | −0.4901 | 0.5951 | 1.933 | −1.0345 | −1.1996 | −0.9378 | −1.8962 | 1.3793 | −0.8242 | −0.7745 | −1.7589 | −0.9362 |
| 79 | −0.2186 | −1.1662 | −1.15 | −0.7646 | −0.3131 | −1.3009 | 1.7065 | −2.237 | −1.5943 | −0.9207 | −0.9506 | 0.7116 | −0.9004 | 0.2218 | −1.887 | −1.096 |
| 80 | 1.0808 | −0.597 | −0.8584 | −0.8226 | −1.4641 | −1.2374 | 0.7571 | −1.4812 | −2.0239 | −0.7141 | −1.4048 | 0.3666 | −0.9214 | −1.3547 | 0.2259 | −1.0798 |
| 81 | −1.3624 | 0.1524 | −1.1858 | −0.8277 | −1.4117 | −1.2232 | −1.2171 | −0.8709 | −0.8319 | −0.8716 | −0.6282 | 1.7031 | −1.7363 | −0.7022 | −1.9667 | −1.0593 |
| 82 | −1.2753 | −0.6617 | −1.2841 | −0.6723 | 0.2354 | 0.0003 | −1.3449 | −0.3184 | −0.8008 | −0.8581 | 0.298 | −1.1678 | −0.6246 | −0.3164 | 1.769 | −0.4109 |
| 83 | −1.0755 | 0.189 | −1.3647 | −0.902 | −1.5986 | −1.9597 | −0.8797 | −1.0385 | −0.5315 | −0.7335 | −0.8141 | 1.275 | −1.888 | −0.2927 | −1.5776 | −1.1293 |
| 84 | −1.8085 | −1.0142 | −1.2811 | −0.9334 | −1.0717 | −0.4323 | 0.9235 | −2.1046 | −1.0034 | −0.9462 | −0.8141 | −1.2629 | −0.9565 | −0.2507 | −1.7703 | −0.6834 |
| 85 | −0.6364 | −1.8746 | −0.8828 | −0.8022 | −1.9914 | 0.1244 | 1.8855 | −1.3999 | −1.2799 | −0.7505 | −0.6868 | −1.4617 | −0.0416 | −1.7382 | −1.2005 | −0.5755 |
| 86 rows × 16 columns. | ||||||||||||||||
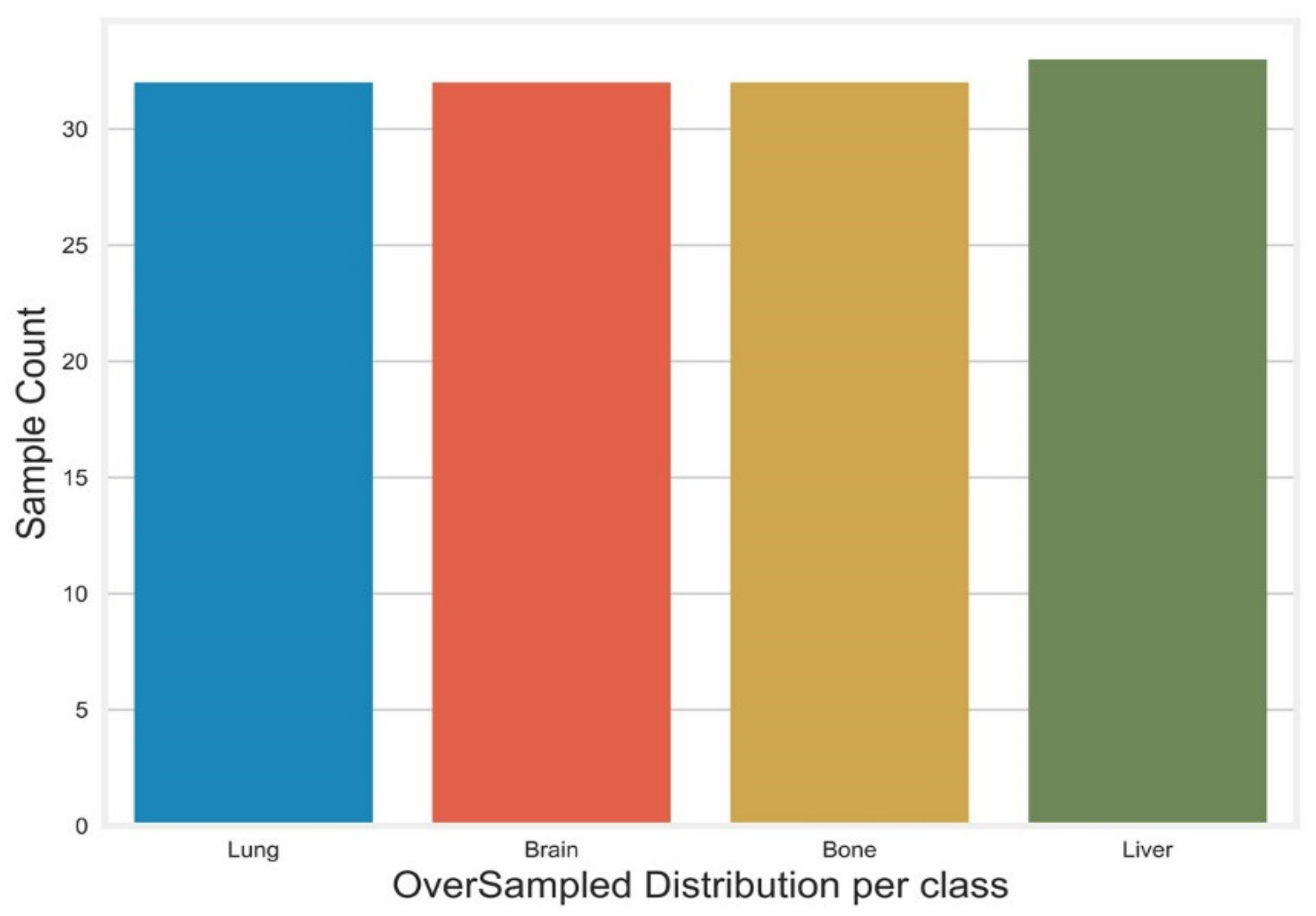
Appendix F. Oversampled Dataset after Applying K-Mean Smote
| 222,865 | 25,973 | 2670 | 347,733 | 4131 | 4283 | 4318 | 441,150 | 51,011 | 6439 | 729,887 | 81,035 | 8120 | 81,575 | 8817 | 90,865 | Metastasis | |
| 0 | 0.198 | 0.9758 | 0.3665 | −0.2696 | 1.0458 | 0.9783 | −0.6444 | 3.782 | 3.171 | 0.3832 | 4.6323 | −0.4311 | −0.2163 | −0.1137 | −0.1295 | 0.7515 | Lung |
| 1 | −0.0865 | 1.1 | 0.7864 | 3.312 | 1.2416 | 0.5305 | 0.9294 | 0.6143 | 0.9198 | 0.1937 | −0.1079 | −0.3811 | 4.3088 | 1.2101 | −0.2767 | −0.4483 | Brain |
| 2 | 1.9646 | 1.1233 | 1.0511 | −0.0854 | 0.9364 | −0.79 | −1.2065 | 0.9356 | 0.1308 | −0.0736 | 2.1 | −0.71 | 0.2795 | 0.9134 | −0.3304 | −0.142 | Brain |
| 3 | −1.192 | −0.6367 | 0.2691 | −0.4562 | 1.7566 | −0.7846 | 1.4329 | −0.7015 | 0.4727 | −0.1349 | −0.7771 | 1.5955 | −0.3288 | 2.6731 | 3.8883 | 1.3303 | Bone |
| 4 | 0.1266 | −0.0604 | 1.7196 | 0.0398 | 1.8735 | −0.659 | 0.0221 | 0.1035 | 0.4401 | −0.0272 | 2.0996 | −0.2556 | −0.4302 | −0.0581 | 0.8987 | −0.5004 | Brain |
| 5 | −0.5906 | 0.3491 | 1.4248 | 0.2006 | 2.0667 | −0.7052 | −0.5249 | −0.2215 | −0.3393 | 0.1466 | −0.8944 | 0.6907 | −0.2445 | −0.2771 | −0.0903 | −0.4262 | Bone |
| 6 | −0.8871 | 0.1041 | 0.1751 | −0.7043 | 1.6466 | −0.3017 | 0.1538 | −0.4897 | 0.4002 | 0.0265 | −0.4333 | 0.4794 | −0.0686 | 1.0238 | −0.2101 | −0.2477 | Bone |
| 7 | 0.4481 | 0.0621 | 0.9934 | 1.1176 | 1.0911 | −0.7203 | 0.0915 | 1.7954 | 1.4472 | −0.0197 | 2.5998 | −0.3057 | −0.5338 | 1.8907 | 0.3507 | −0.4034 | Brain |
| 8 | −0.2063 | 1.0118 | 0.0792 | −0.7138 | 1.0369 | 1.4341 | 1.8265 | −0.2083 | 0.2059 | −0.0171 | −0.8552 | 1.9019 | −0.5076 | 0.847 | 0.2876 | 0.1463 | Bone |
| 9 | −0.2731 | 1.155 | 2.0704 | 1.0202 | 2.5062 | −0.6621 | −0.5338 | −0.1218 | 0.7685 | 0.1553 | 0.1663 | −0.5509 | −0.2045 | 1.5289 | 0.1918 | −0.2485 | Brain |
| … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … | … |
| 119 | −1.2187 | −2.5334 | −1.328 | 1.6502 | −0.1868 | 2.3117 | −0.7413 | −1.161 | −0.9107 | −0.8275 | −0.8525 | −0.9858 | −1.3184 | −1.3744 | −1.9571 | 0.8591 | Liver |
| 120 | −0.9832 | −1.4084 | −1.022 | 0.3152 | −1.2142 | 1.989 | −0.7787 | −1.2526 | −0.7445 | −0.5679 | −0.9831 | −1.2853 | −1.8635 | −1.9452 | −1.5483 | 3.7285 | Liver |
| 121 | −1.4467 | −1.1812 | −1.3396 | 0.4043 | −0.9398 | 1.6933 | −1.1969 | −1.693 | −0.9057 | −0.8883 | −1.0399 | −1.4553 | −1.4607 | −2.0827 | −2.2189 | 0.1661 | Liver |
| 122 | −0.5328 | −1.365 | −0.9757 | 1.0245 | −0.9261 | 2.2429 | −0.9658 | −1.1536 | −0.8336 | −0.5485 | −0.9617 | −1.2656 | −1.912 | −2.0633 | −1.2153 | 3.7183 | Liver |
| 123 | −0.7801 | −1.3888 | −1.0011 | 0.6352 | −1.0842 | 2.1035 | −0.8631 | −1.2079 | −0.7847 | −0.5592 | −0.9735 | −1.2764 | −1.8854 | −1.9984 | −1.398 | 3.7239 | Liver |
| 124 | −1.1877 | −0.5688 | −1.1857 | −0.0131 | −1.3437 | 1.6019 | −1.3146 | −1.7328 | −0.8642 | −0.7686 | −1.1032 | −1.614 | −1.7331 | −2.4309 | −1.9283 | 1.4033 | Liver |
| 125 | −1.331 | −2.2994 | −1.2769 | 1.0944 | −0.5203 | 2.1472 | −0.6812 | −1.2176 | −0.8412 | −0.7769 | −0.8893 | −1.0595 | −1.4218 | −1.458 | −1.988 | 1.5002 | Liver |
| 126 | −1.0673 | −2.7155 | −1.3628 | 2.193 | 0.1203 | 2.4782 | −0.815 | −1.1022 | −0.9786 | −0.8655 | −0.8199 | −0.9239 | −1.2419 | −1.3232 | −1.8872 | 0.3424 | Liver |
| 127 | −1.5902 | −0.5177 | −1.3223 | −0.3542 | −1.3972 | 1.3651 | −1.3603 | −1.9386 | −0.8735 | −0.8917 | −1.1338 | −1.6824 | −1.5646 | −2.4134 | −2.3412 | 0.1555 | Liver |
| 128 | −1.7172 | −1.307 | −1.126 | −0.8419 | −1.6835 | 1.5209 | −0.5981 | −1.5013 | −0.6315 | −0.638 | −1.0401 | −1.3814 | −1.7628 | −1.8607 | −2.1368 | 3.2783 | Liver |
| 129 rows × 17 columns. | |||||||||||||||||
References
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- JPMA—Journal of Pakistan Medical Association. Available online: https://jpma.org.pk/article-details/1863 (accessed on 13 February 2021).
- Menhas, R.; Umer, S. Breast Cancer among Pakistani Women. Iran. J. Public Health 2015, 44, 586–587. [Google Scholar] [PubMed]
- Zaheer, S.; Shah, N.; Maqbool, S.A.; Soomro, N.M. Estimates of Past and Future Time Trends in Age-Specific Breast Cancer Incidence among Women in Karachi, Pakistan: 2004–2025. BMC Public Health 2019, 19, 1001. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.R.; Varadhachary, G.R.; Taylor, S.H.; Wei, W.; Raber, M.N.; Lenzi, R.; Abbruzzese, J.L. Metastatic Patterns in Adenocarcinoma. Cancer 2006, 106, 1624–1633. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast Cancer Subtypes Predict the Preferential Site of Distant Metastases: A SEER Based Study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [Green Version]
- Schlappack, O.K.; Baur, M.; Steger, G.; Dittrich, C.; Moser, K. The Clinical Course of Lung Metastases from Breast Cancer. Klin. Wochenschr. 1988, 66, 790–795. [Google Scholar] [CrossRef]
- Xiao, W.; Zheng, S.; Liu, P.; Zou, Y.; Xie, X.; Yu, P.; Tang, H.; Xie, X. Risk Factors and Survival Outcomes in Patients with Breast Cancer and Lung Metastasis: A Population-Based Study. Cancer Med. 2018, 7, 922–930. [Google Scholar] [CrossRef]
- GSE14020—NCBI. Available online: https://www.ncbi.nlm.nih.gov/search/all/?term=GSE14020 (accessed on 6 December 2020).
- GSE54323—NCBI. Available online: https://www.ncbi.nlm.nih.gov/search/all/?term=GSE54323 (accessed on 6 December 2020).
- Daoud, M.; Mayo, M. A Survey of Neural Network-Based Cancer Prediction Models from Microarray Data. Artif. Intell. Med. 2019, 97, 204–214. [Google Scholar] [CrossRef]
- Yazici, H.; Akin, B. Molecular Genetics of Metastatic Breast Cancer. In Tumour Progression and Metastasis; 2020; Available online: https://books.google.com.hk/books?hl=zh-CN&lr=&id=WXL8DwAAQBAJ&oi=fnd&pg=PA33&dq=Molecular+Genetics+of+Metastatic+Breast+Cancer.+In+Tumou&ots=fD07Myo0Zn&sig=N7UQpRfEosuIQxpTXI4KZx755Yc&redir_esc=y&hl=zh-CN&sourceid=cndr#v=onepage&q=Molecular%20Genetics%20of%20Metastatic%20Breast%20Cancer.%20In%20Tumou&f=false (accessed on 10 June 2022).
- Saunus, J.M.; Momeny, M.; Simpson, P.T.; Lakhani, S.R.; Da Silva, L. Molecular Aspects of Breast Cancer Metastasis to the Brain. Genet. Res. Int. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Mu, P. Targeting Breast Cancer Metastasis. Breast Cancer Basic Clin. Res. 2015, 9, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Ma, R.; Feng, Y.; Lin, S.; Chen, J.; Lin, H.; Liang, X.; Zheng, H.; Cai, X. Mechanisms Involved in Breast Cancer Liver Metastasis. J. Transl. Med. 2015, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.Y.; Gong, Y.; Ye, F.G.; Ling, H.; Hu, X. Incidence and Prognostic Factors of Patients with Synchronous Liver Metastases upon Initial Diagnosis of Breast Cancer: A Population-Based Study. Cancer Manag. Res. 2018, 10, 5937–5950. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, R.M.S.M.; Mustafa, D.A.; Soffietti, R.; Kros, J.M. Breast Cancer Brain Metastasis: Molecular Mechanisms and Directions for Treatment. Neuro. Oncol. 2018, 20, 1439–1449. [Google Scholar] [CrossRef]
- Brosnan, E.M.; Anders, C.K. Understanding Patterns of Brain Metastasis in Breast Cancer and Designing Rational Therapeutic Strategies. Ann. Transl. Med. 2018, 6, 163. [Google Scholar] [CrossRef]
- Stella, G.M.; Kolling, S.; Benvenuti, S.; Bortolotto, C. Lung-Seeking Metastases. Cancers 2019, 11, 1010. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast Cancer Lung Metastasis: Molecular Biology and Therapeutic Implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Affimetrix Human Genome U133 Arrays the Most Comprehensive Coverage of the Human Genome in Two Flexible Formats: Single-Array Cartridges and Multi-Array Plates; 2017; Available online: https://www.thermofisher.com/ (accessed on 1 January 2022).
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-Centered Information at NCBI. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef]
- SOFT—GEO—NCBI. Available online: https://www.ncbi.nlm.nih.gov/geo/info/soft.html (accessed on 2 January 2021).
- GEOparse—GEOparse 1.2.0 Documentation. Available online: https://geoparse.readthedocs.io/en/latest/introduction.html (accessed on 19 December 2020).
- Liew, A.W.-C.; Law, N.-F.; Yan, H. Missing Value Imputation for Gene Expression Data: Computational Techniques to Recover Missing Data from Available Information. Brief. Bioinform. 2011, 12, 498–513. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorso, G. Machine Learning Algorithms: A Reference Guide to Popular Algorithms for Data Science and Machine Learning; Packt Publishing: Birmingham, UK, 2017; ISBN 1785889621. [Google Scholar]
- Lin, W.C.; Tsai, C.F. Missing Value Imputation: A Review and Analysis of the Literature (2006–2017). Artif. Intell. Rev. 2020, 53, 1487–1509. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing Value Estimation Methods for DNA Microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, T.; Tibshirani, R.; Sherlock, G.; Eisen, M.; Brown, P.; Botstein, D. Imputing Missing Data for Gene Expression Arrays. Stanford Univ. Stat. Dep. Tech. Rep. 2006, 3, 27. Available online: http://www.stat.stanford.edu/Hast.pdf/cll/qxd. (accessed on 2 January 2021).
- Sklearn.Impute.KNNImputer—Scikit-Learn 0.24.0 Documentation. Available online: https://scikit-learn.org/stable/modules/generated/sklearn.impute.KNNImputer.html (accessed on 8 January 2021).
- Dash, R.; Misra, B.B. Performance Analysis of Clustering Techniques over Microarray Data: A Case Study. Phys. A Stat. Mech. Its Appl. 2018, 493, 162–176. [Google Scholar] [CrossRef]
- Mukaka, M.M. Statistics Corner: A Guide to Appropriate Use of Correlation Coefficient in Medical Research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar] [PubMed]
- Darst, B.F.; Malecki, K.C.; Engelman, C.D. Using Recursive Feature Elimination in Random Forest to Account for Correlated Variables in High Dimensional Data. BMC Genet. 2018, 19, 65. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xie, W.; Liu, T. Efficient Feature Selection and Classification for Microarray Data. PLoS ONE 2018, 13, 1–21. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority Over-Sampling Technique. J. Artif. Int. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Douzas, G.; Bacao, F.; Last, F. Improving Imbalanced Learning through a Heuristic Oversampling Method Based on K-Means and {SMOTE}. Inf. Sci. (NY) 2018, 465, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Riaz, F. Integration-of-Machine-Learning-and-Microarrays-for-the-Identification-of-Breast-Cancer-in-Vital-Org/Oversampled_afterKmeanSmote_data.csv at Master Faisalriazz/Integration-of-Machine-Learning-and-Microarrays-for-the-Identification-of-Breast-Cancer-in-Vi. Available online: https://github.com/faisalriazz/Integration-of-Machine-Learning-and-Microarrays-for-the-Identification-of-Breast-Cancer-in-Vital-Org/blob/master/oversampled_afterKmeanSmote_data.csv (accessed on 16 July 2021).
- Ritu, A. Latiyan Shiwam Prediction of Breast Cancer Using Different Machine Learning Algorithms. In Proceedings of 6th International Conference on Recent Trends in Computing; Mahapatra, R.P., Panigrahi, B.K., Kaushik, B.K., Roy, S., Eds.; Springer Nature Pte. Ltd.: Berlin, Germany, 2021; pp. 369–383. ISBN 978-981-334-501-0. [Google Scholar]
- Rajaguru, H.; Sannasi Chakravarthy, S.R. Analysis of Decision Tree and K-Nearest Neighbor Algorithm in the Classification of Breast Cancer. Asian Pacific. J. Cancer Prev. 2019, 20, 3777–3781. [Google Scholar] [CrossRef] [Green Version]
- Al-Salihy, N.K.; Ibrikci, T. Classifying Breast Cancer by Using Decision Tree Algorithms. ACM Int. Conf. Proc. Ser. 2017, 144–148. [Google Scholar] [CrossRef]
- Nel, I.; Morawetz, E.W.; Tschodu, D.; Käs, J.A.; Aktas, B. The Mechanical Fingerprint of Circulating Tumour Cells (Ctcs) in Breast Cancer Patients. Cancers 2021, 13, 1119. [Google Scholar] [CrossRef]
- Andreas, C.M.; Sarah, G. Introduction to Machine Learning with Python. In Introduction to Machine Learning with Python; O’Reilly Media, Inc.: Sevastopol, CA, USA, 2016; pp. 83–84. ISBN 9781449369415. [Google Scholar]
- Huang, S.; Nianguang, C.A.I.; Penzuti Pacheco, P.; Narandes, S.; Wang, Y.; Wayne, X.U. Applications of Support Vector Machine (SVM) Learning in Cancer Genomics. Cancer Genom. Proteom. 2018, 15, 41–51. [Google Scholar] [CrossRef] [Green Version]





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Platform | No. of Samples |
|---|---|---|
| GSE14020 | GPL96 | 36 |
| GPL570 | 29 | |
| GSE54323 | GPL570 | 29 |
| Algorithm | Pros | Cons |
|---|---|---|
| KNN | Very easy to understand and implement. | Lazy learning algorithm. |
| Does not make any assumption about data. | Poor performance with high dimension dataset. | |
| It changes to accommodate the new data points when exposed to new data. | Data scaling is required. | |
| DT | Data scaling or normalization is not required. | Prone to overfitting the model. |
| Missing values does not have considerable impact. | Training time is higher. | |
| Easy to interpret and visualize. | Sensitivity to data changes is quite high. A small change can affect the result significantly. | |
| RF | Random Forest is an ensemble based on decision trees. It ensures the reduction in overall variance and error. | Not easy to interpret. |
| Performs well with higher dimensions. | Tuning of hyperparameters is required to improve performance. | |
| Missing values and outliers do not have considerable impact. | Training time is lower but prediction time is higher. | |
| Not prone to overfitting. | ||
| SVM | Provides high accuracy and performance in higher dimensional data. | Execution time is higher for larger dataset. |
| Most suitable algorithm when classes are separable either linear or non-linear. | Performance degrades in case of non-separable classes. | |
| Susceptibility to outliers is low. | Hyperparameter optimization is required for better generalized performance. |
| Classifier | Class | Precision | Recall | F1 Score | PR-AUC | ROC-AUC |
|---|---|---|---|---|---|---|
| DT | Lung | 0.88 | 0.70 | 0.78 | 0.76 | 0.87 |
| Brain | 1.00 | 0.89 | 0.94 | 1.00 | 0.90 | |
| Bone | 0.90 | 0.90 | 0.90 | 0.54 | 0.96 | |
| Liver | 0.77 | 1.00 | 0.87 | 0.70 | 0.95 | |
| AVG Precision = 0.77 | AVG ROC-AUC = 0.92 | |||||
| RF | Lung | 1.00 | 0.90 | 0.95 | 0.98 | 1.00 |
| Brain | 0.90 | 0.80 | 0.95 | 1.00 | 1.00 | |
| Bone | 0.89 | 1.00 | 0.84 | 0.90 | 0.98 | |
| Liver | 0.82 | 0.90 | 0.86 | 1.00 | 1.00 | |
| AVG Precision = 0.96 | AVG ROC-AUC = 1.00 | |||||
| KNN | Lung | 0.89 | 0.80 | 0.84 | 0.98 | 1.00 |
| Brain | 0.90 | 1.00 | 0.95 | 1.00 | 1.00 | |
| Bone | 1.00 | 0.70 | 0.82 | 0.95 | 0.99 | |
| Liver | 0.76 | 1.00 | 0.87 | 0.98 | 0.99 | |
| AVG Precision = 0.96 | AVG ROC-AUC = 1.00 | |||||
| SVM | Lung | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Brain | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| Bone | 0.91 | 1.00 | 0.95 | 0.97 | 0.99 | |
| Liver | 1.00 | 0.99 | 0.99 | 0.97 | 1.00 | |
| AVG Precision = 0.99 | AVG ROC-AUC = 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riaz, F.; Abid, F.; Din, I.U.; Kim, B.-S.; Almogren, A.; Durar, S.U. Identification of Secondary Breast Cancer in Vital Organs through the Integration of Machine Learning and Microarrays. Electronics 2022, 11, 1879. https://doi.org/10.3390/electronics11121879
Riaz F, Abid F, Din IU, Kim B-S, Almogren A, Durar SU. Identification of Secondary Breast Cancer in Vital Organs through the Integration of Machine Learning and Microarrays. Electronics. 2022; 11(12):1879. https://doi.org/10.3390/electronics11121879
Chicago/Turabian StyleRiaz, Faisal, Fazeel Abid, Ikram Ud Din, Byung-Seo Kim, Ahmad Almogren, and Shajara Ul Durar. 2022. "Identification of Secondary Breast Cancer in Vital Organs through the Integration of Machine Learning and Microarrays" Electronics 11, no. 12: 1879. https://doi.org/10.3390/electronics11121879