
Substituent Control of Near-Infrared Absorption of Triphenylamine Radical Cation

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Theoretical Calculations
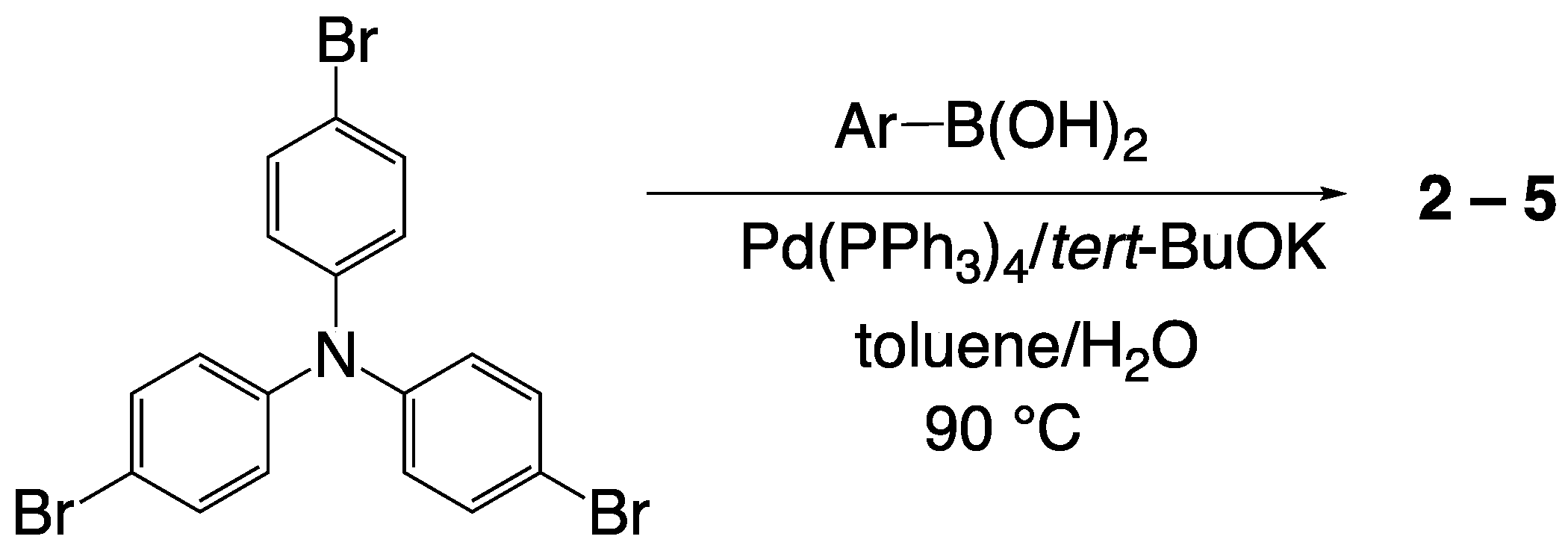
2.2. Synthesis
2.3. Solubility
2.4. Cyclic Voltammetry Measurements
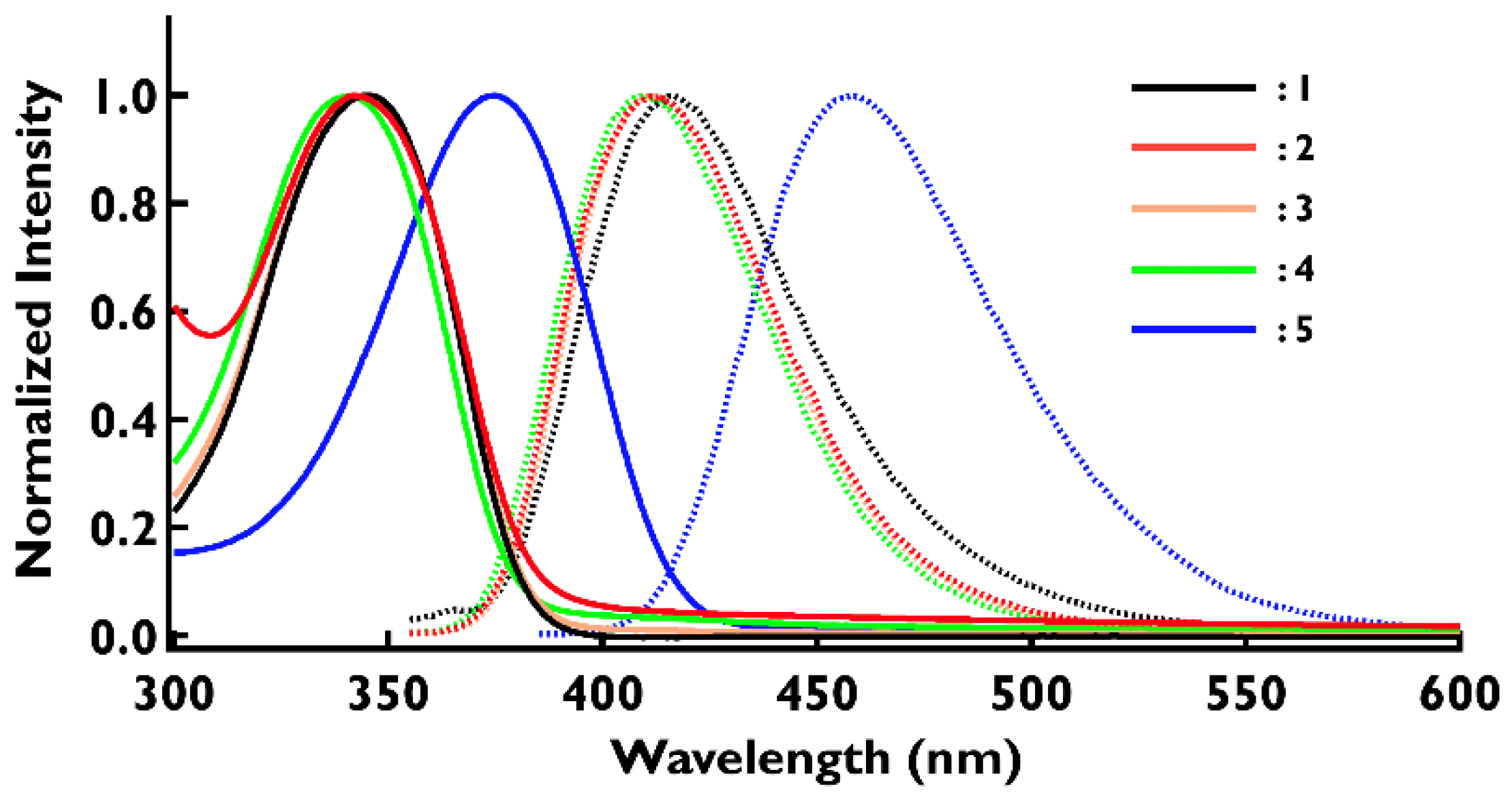
2.5. Absorption and Fluorescence Spectra of the Neutral Species, 1–5
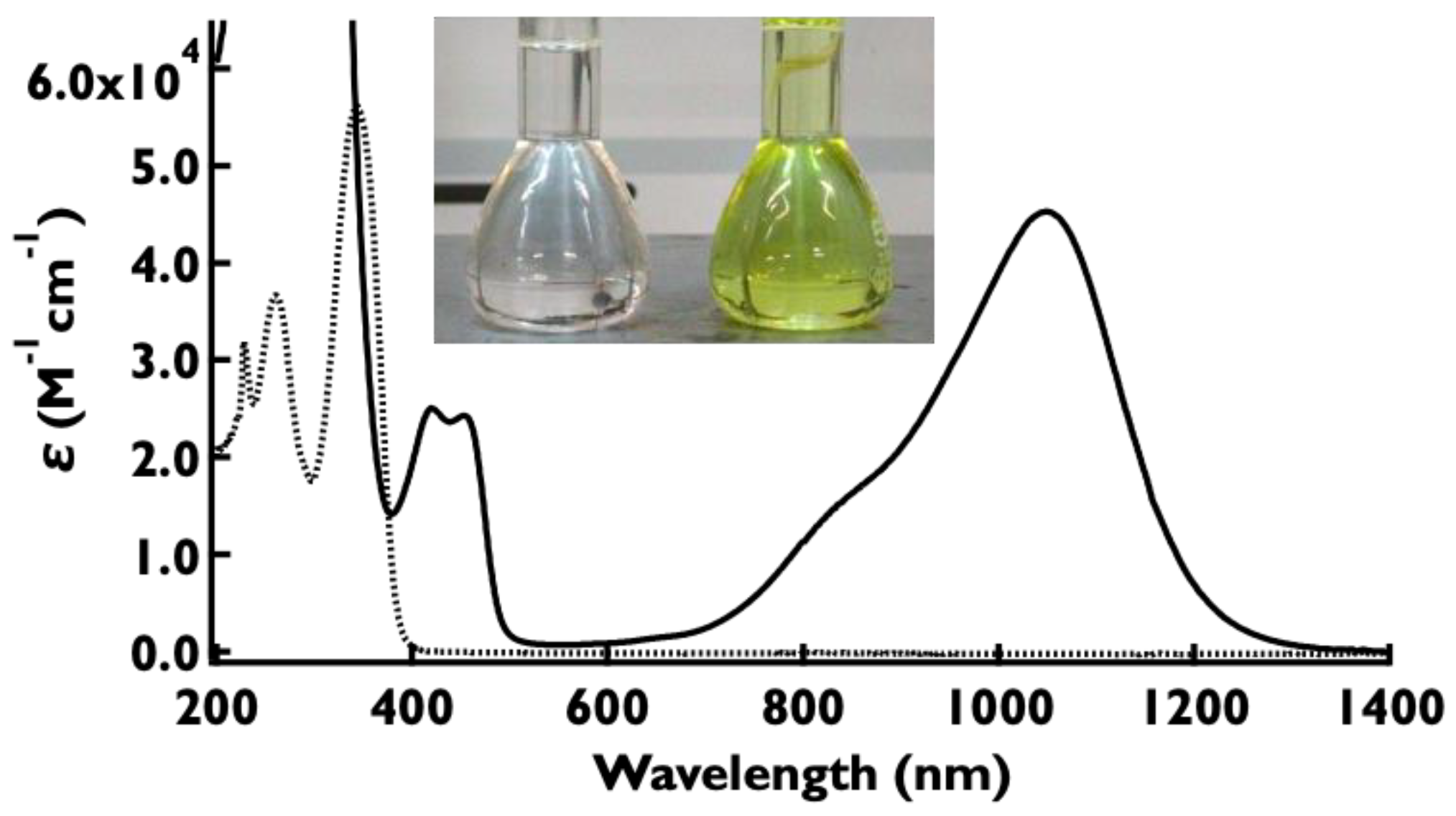
2.6. UV–Vis–NIR Absorption Spectra of the Oxidized Species, 1•+–5•+
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, S.-H.; Cheong, H.M.; Zhang, J.-G.; Mascarenhas, A.; Benson, D.K.; Deb, S.K. Electrochromic mechanism in a-WO3−y thin films. Appl. Phys. Lett. 1999, 74, 242–244. [Google Scholar] [CrossRef]
- Deb, S.K. Optical and photoelectric properties and colour centres in thin films of tungsten oxide. Philos. Mag. 1973, 27, 801–822. [Google Scholar] [CrossRef]
- Fabian, J.; Nakazumi, H.; Matsuoka, M. Near-infrared absorbing dyes. Chem. Rev. 1992, 92, 1197–1226. [Google Scholar] [CrossRef]
- Rao, R.S.; Suman; Singh, S.P. Near-Infrared (>1000 nm) Light-Harvesters: Design, Synthesis and Applications. Chem.—Eur. J. 2020, 26, 16582–16593. [Google Scholar] [CrossRef]
- Li, L.; Dong, X.; Li, J.; Wei, J. A short review on NIR-II organic small molecule dyes. Dye. Pigment. 2020, 183, 108756. [Google Scholar] [CrossRef]
- Li, B.; Zhao, M.; Zhang, F. Rational Design of Near-Infrared-II Organic Molecular Dyes for Bioimaging and Biosensing. ACS Mater. Lett. 2020, 2, 905–917. [Google Scholar] [CrossRef]
- Qi, J.; Qiao, W.; Wang, Z.Y. Advances in Organic Near-Infrared Materials and Emerging Applications. Chem. Rec. 2016, 16, 1531–1548. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, J. Higher order acenes and fused acenes with near-infrared absorption and emission. Aust. J. Chem. 2011, 64, 519–528. [Google Scholar] [CrossRef]
- Wang, Z. Near-Infrared Organic Materials and Emerging Applications, 1st ed.; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Seo, E.T.; Nelson, R.F.; Fritsch, J.M.; Marcoux, L.S.; Leedy, D.W.; Adams, R.N. Anodic Oxidation Pathways of Aromatic Amines. Electrochemical and Electron Paramagnetic Resonance Studies. J. Am. Chem. Soc. 1966, 88, 3498–3503. [Google Scholar] [CrossRef]
- Nelson, R.R.; Adams, R.N. Anodic oxidation pathways of substituted triphenylamines. II. Quantitative studies of benzidine formation. J. Am. Chem. Soc. 1968, 90, 3925–3930. [Google Scholar] [CrossRef]
- Nelson, R.F.; Philp, R.H. Electrochemical and spectroscopic studies of cation radicals. 4. Stopped-flow determination of triarylaminium radical coupling rate constants. J. Phys. Chem. 1979, 83, 713–716. [Google Scholar] [CrossRef]
- Yano, M.; Tamada, K.; Nakai, M.; Mitsudo, K.; Kashiwagi, Y. Near-Infrared Absorbing Molecule Based on Triphenylamine Radical Cation with Extended Homoaryl π-System. Colorants 2022, 1, 226–235. [Google Scholar] [CrossRef]
- Yano, M.; Inada, Y.; Hayashi, Y.; Yajima, T.; Mitsudo, K.; Kashiwagi, Y. Photo- and Redox-active Benzofuran-appended Triphenylamine and Near-infrared Absorption of Its Radical Cation. Chem. Lett. 2020, 49, 685–688. [Google Scholar] [CrossRef]
- Yano, M.; Inada, Y.; Hayashi, Y.; Nakai, M.; Mitsudo, K.; Kashiwagi, Y. Near-infrared absorption of a benzothiophene-appended triphenylamine radical cation: A novel molecular design of NIR-II dye. Dye. Pigment. 2022, 197, 109929. [Google Scholar] [CrossRef]
- Chen, C.; Zheng, Q.; Ni, S.; Wang, H. Facile one-pot nanocatalysts encapsulation of palladium-NHC complexes for aqueous Suzuki-Miyaura couplings. New J. Chem. 2018, 42, 4624–4630. [Google Scholar] [CrossRef]
- Rao, M.L.N.; Banerjee, D.; Dhanorkar, R.J. Pd-catalyzed coupling of aryl iodides with triarylbismuths as atom-economic multi-coupling organometallic nucleophiles under mild conditions. Tetrahedron Lett. 2010, 51, 6101–6104. [Google Scholar] [CrossRef]
- Hu, B.; Chen, X.; Wang, Y.; Lu, P.; Wang, Y. Structure-Property Investigations of Substituted Triarylamines and Their Applications as Fluorescent pH Sensors. Chem.—Asian J. 2013, 8, 1144–1151. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, T.; Chen, X.; Chen, Q.; Xue, P. Spacer group-controlled luminescence and response of C 3-symmetric triphenylamine derivatives towards force stimuli. CrystEngComm 2021, 23, 202–209. [Google Scholar] [CrossRef]
- Kwon, J.; Kim, M.K.; Hong, J.-P.; Lee, W.; Noh, S.; Lee, C.; Lee, S.; Hong, J.-I. 4,4′,4″-Tris(4-naphthalen-1-yl-phenyl)amine as a multifunctional material for organic light-emitting diodes, organic solar cells, and organic thin-film transistors. Org. Electron. 2010, 11, 1288–1295. [Google Scholar] [CrossRef]
- Kwon, J.; Kim, M.K.; Hong, J.-P.; Lee, W.; Lee, S.; Hong, J.-I. A Multifunctional Material Based on Triphenylamine and a Naphthyl Unit for Organic Light-Emitting Diodes, Organic Solar Cells, and Organic Thin-Film Transistors. Bull. Korean Chem. Soc. 2013, 34, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Rim, Y.S.; Bae, S.; Chen, H.; De Marco, N.; Yang, Y. Recent Progress in Materials and Devices toward Printable and Flexible Sensors. Adv. Mater. 2016, 28, 4415–4440. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Mitsui, C.; Yamagishi, M.; Nakahara, K.; Soeda, J.; Hirose, Y.; Miwa, K.; Sato, H.; Yamano, A.; Matsushita, T.; et al. V-shaped organic semiconductors with solution processability, high mobility, and high thermal durability. Adv. Mater. 2013, 25, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1380. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E0 (V vs. Fc/Fc+) | EHOMO (eV) |
|---|---|---|
| 1 | 0.42 | −4.97 |
| 2 | 0.33 | −4.81 |
| 3 | 0.37 | −4.91 |
| 4 | 0.46 | −5.00 |
| 5 | 0.60 | −5.25 |
| Compound | Absorption Spectra | Fluorescence Spectra | ||
|---|---|---|---|---|
| Obsd. | Calcd. | Obsd. | ||
| λmax (nm) | log ε | λmax (nm) | λmax (nm) | |
| 1 | 344 | 4.78 | 363 | 416 |
| 2 | 343 | 4.75 | 362 | 412 |
| 3 | 345 | 4.83 | 364 | 411 |
| 4 | 341 | 4.64 | 361 | 410 |
| 5 | 375 | 4.86 | 416 | 459 |
| Absorption Spectra | Energy Levels Calculated by DFT | |||||
|---|---|---|---|---|---|---|
| Obsd. | Calcd. | HOMO (eV) | SOMO (eV) | ΔE (eV) | ||
| λmax (nm) | log ε | λmax (nm) | ||||
| 1•+ | 862 | 4.34 | 856 | −6.75 | −4.84 | 1.91 |
| 2•+ | 1053 | 4.65 | 1109 | −6.10 | −4.64 | 1.46 |
| 3•+ | 925 | 4.78 | 931 | −6.52 | −4.76 | 1.76 |
| 4•+ | 870 | 4.56 | 891 | −6.68 | −4.86 | 1.82 |
| 5•+ | 820 | 4.49 | 815 | −7.13 | −5.13 | 2.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yano, M.; Sasaoka, M.; Tamada, K.; Nakai, M.; Yajima, T.; Mitsudo, K.; Kashiwagi, Y. Substituent Control of Near-Infrared Absorption of Triphenylamine Radical Cation. Colorants 2022, 1, 354-362. https://doi.org/10.3390/colorants1030021
Yano M, Sasaoka M, Tamada K, Nakai M, Yajima T, Mitsudo K, Kashiwagi Y. Substituent Control of Near-Infrared Absorption of Triphenylamine Radical Cation. Colorants. 2022; 1(3):354-362. https://doi.org/10.3390/colorants1030021
Chicago/Turabian StyleYano, Masafumi, Mai Sasaoka, Kohei Tamada, Misaki Nakai, Tatsuo Yajima, Koichi Mitsudo, and Yukiyasu Kashiwagi. 2022. "Substituent Control of Near-Infrared Absorption of Triphenylamine Radical Cation" Colorants 1, no. 3: 354-362. https://doi.org/10.3390/colorants1030021