Effect of Cellulose Microfiber Silylation Procedures on the Properties and Antibacterial Activity of Polydimethylsiloxane

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Treatment of Cellulose Microfiber Surfaces
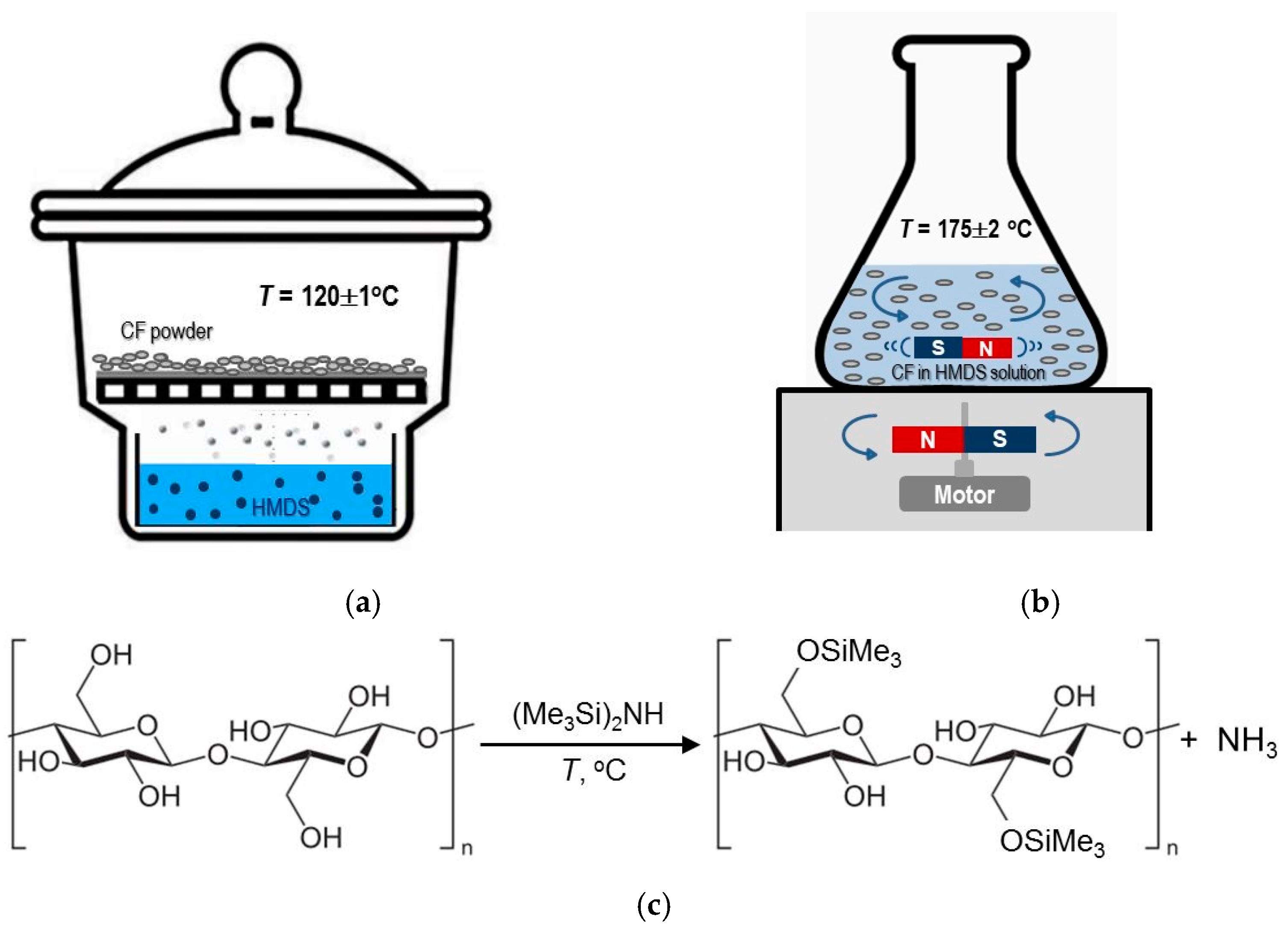
2.2.1. Silylation Methods
- Method I
- Method II
2.2.2. Preparation of CF–Ag Nanohybrid
2.3. Preparation of Composites
2.4. Characterization
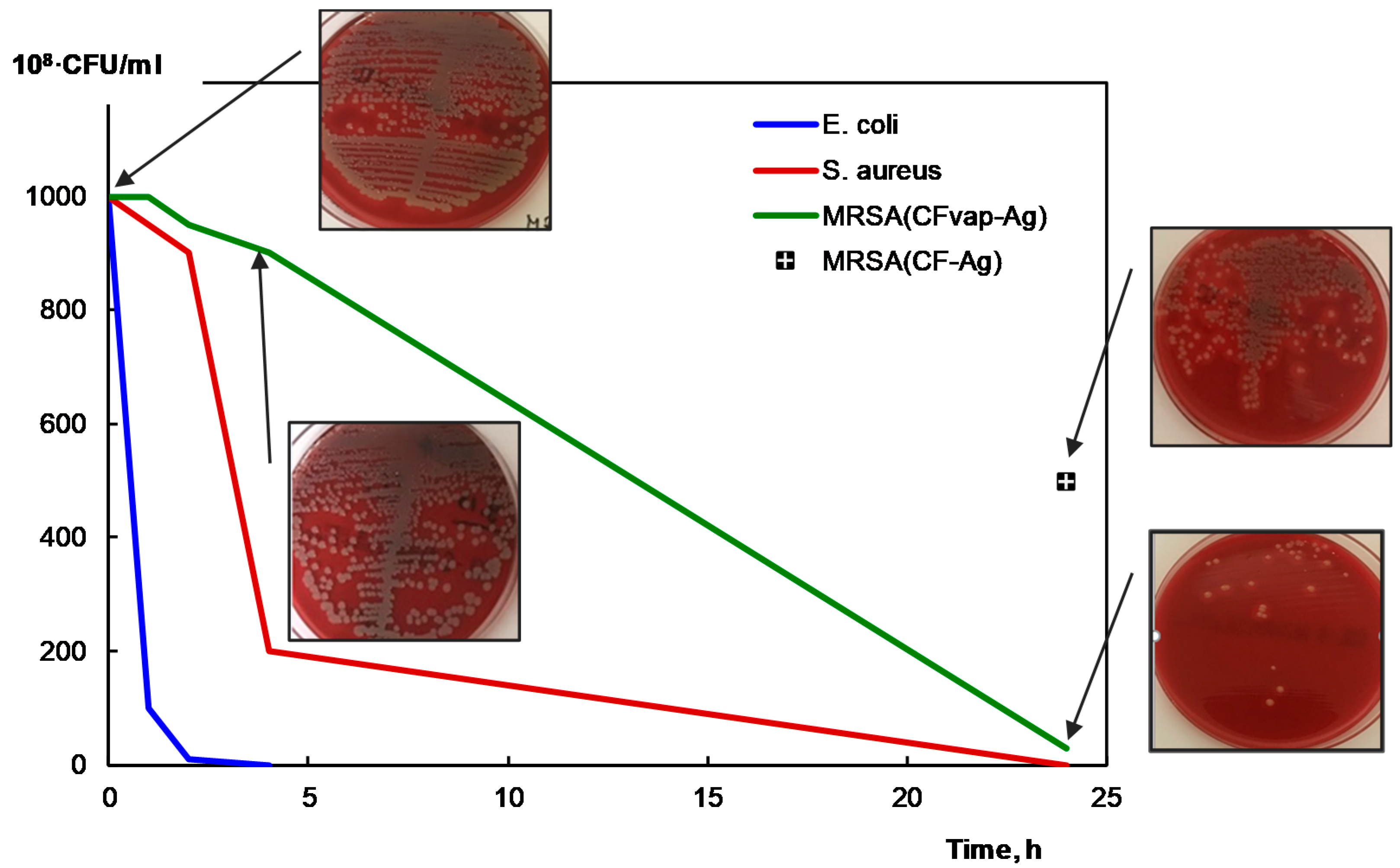
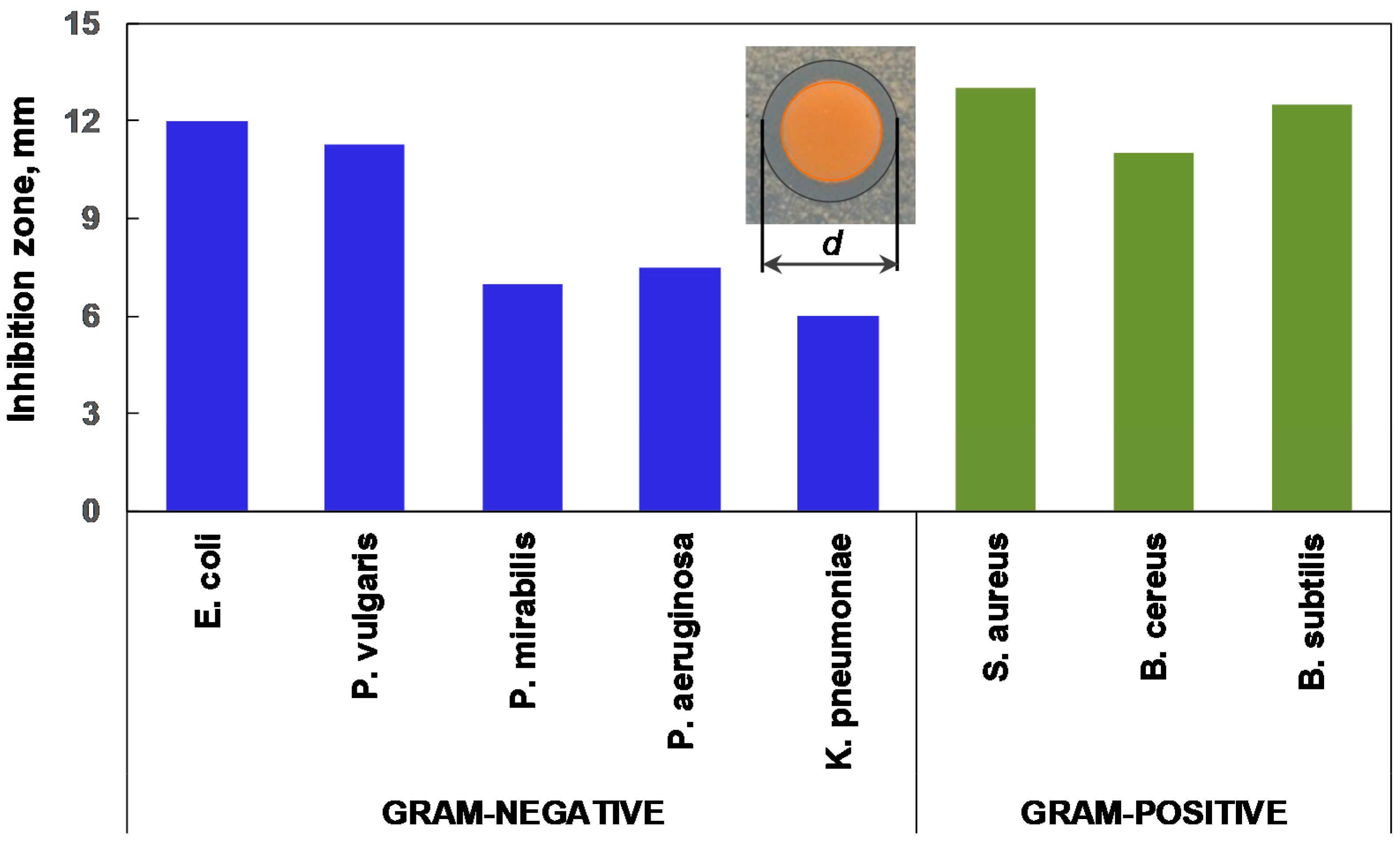
2.5. Antibacterial Activity Studies
3. Results
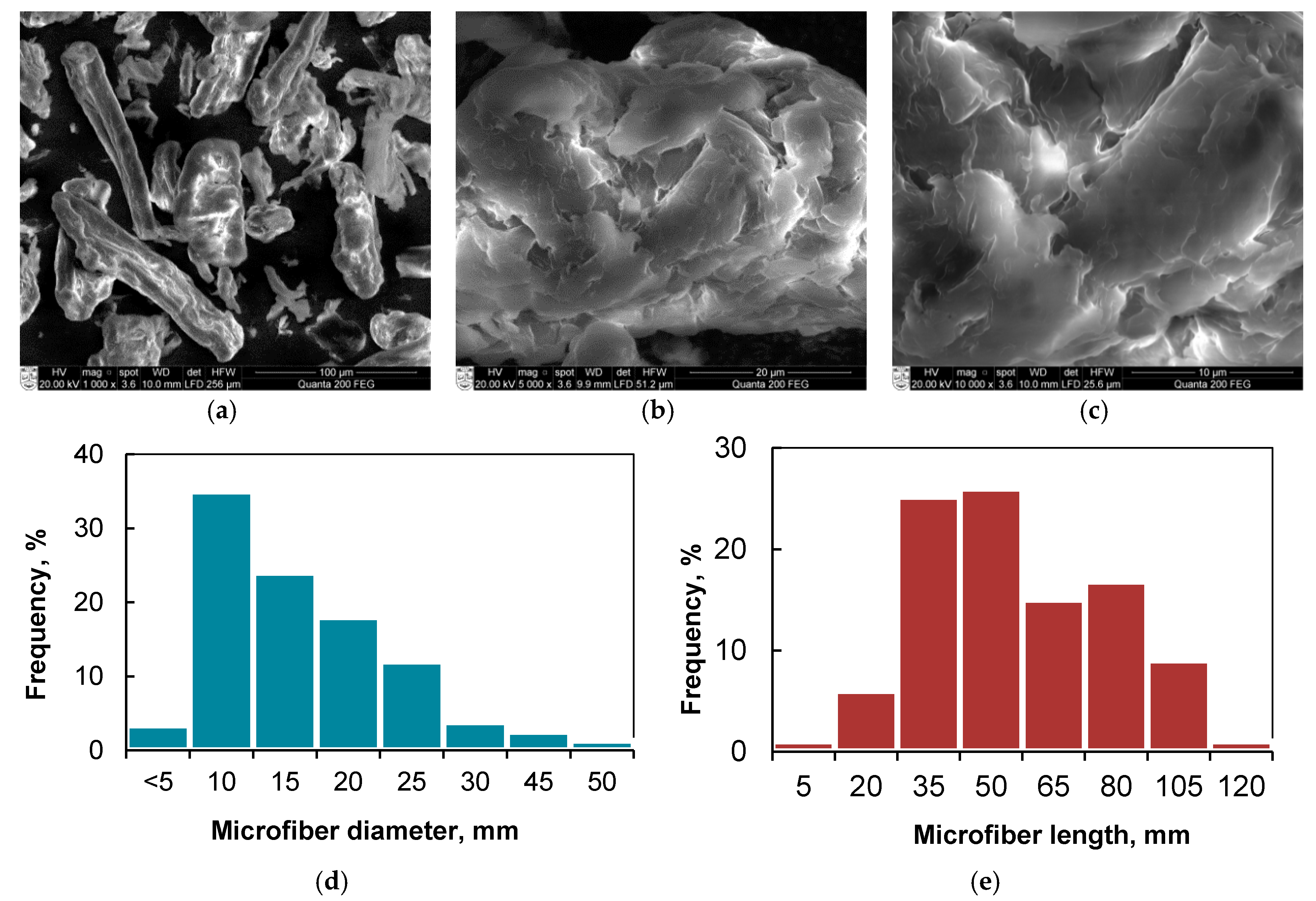
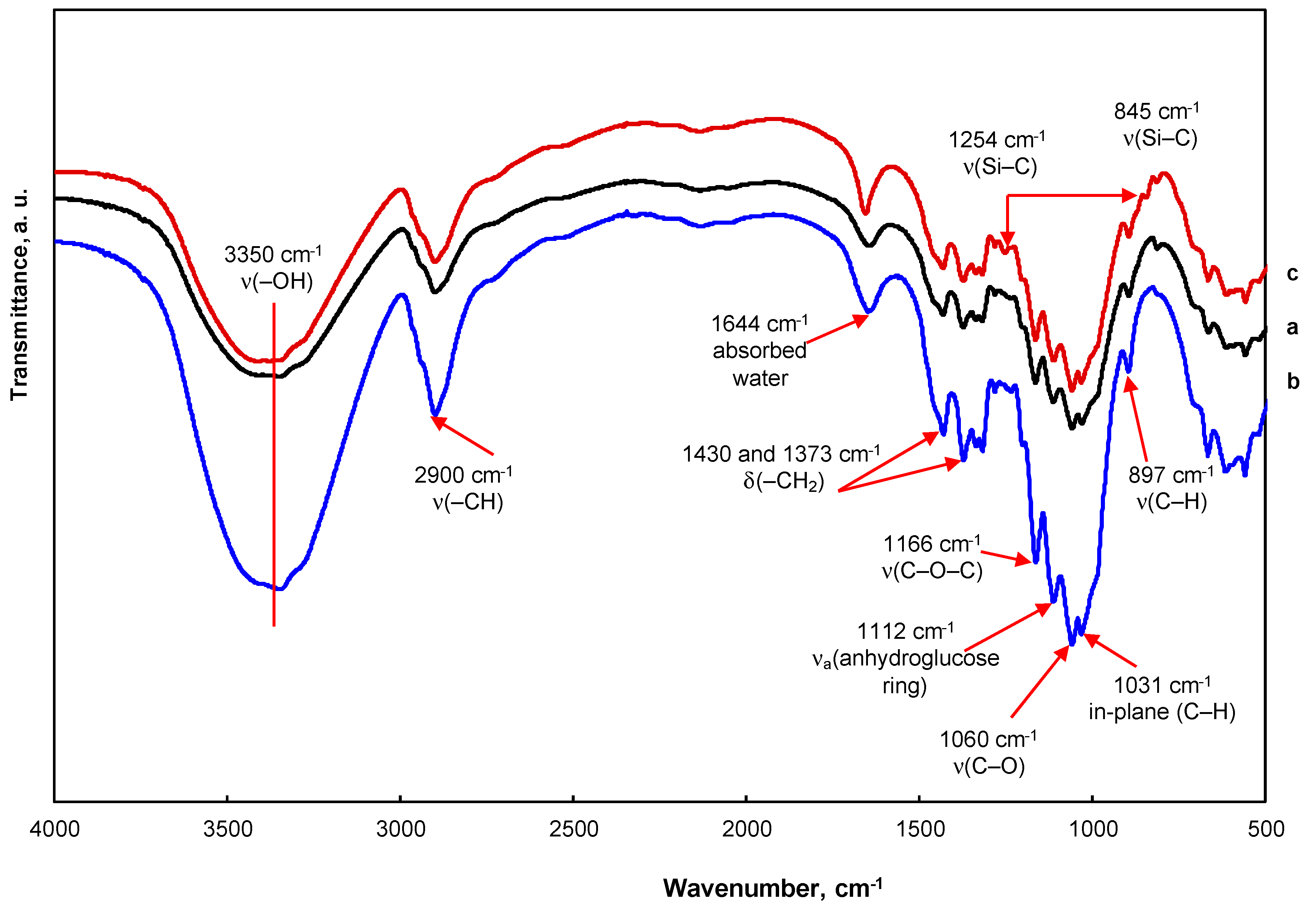
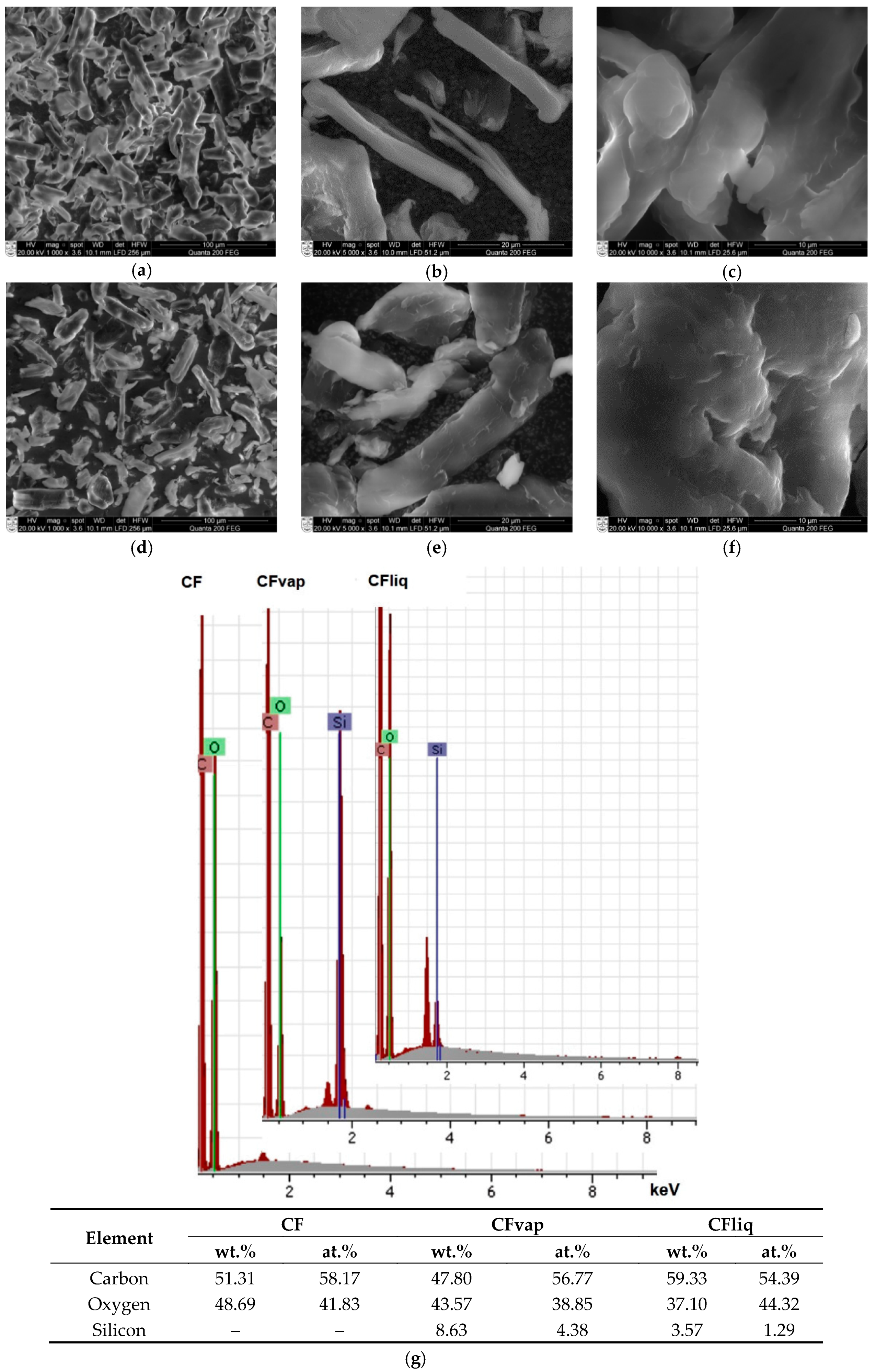
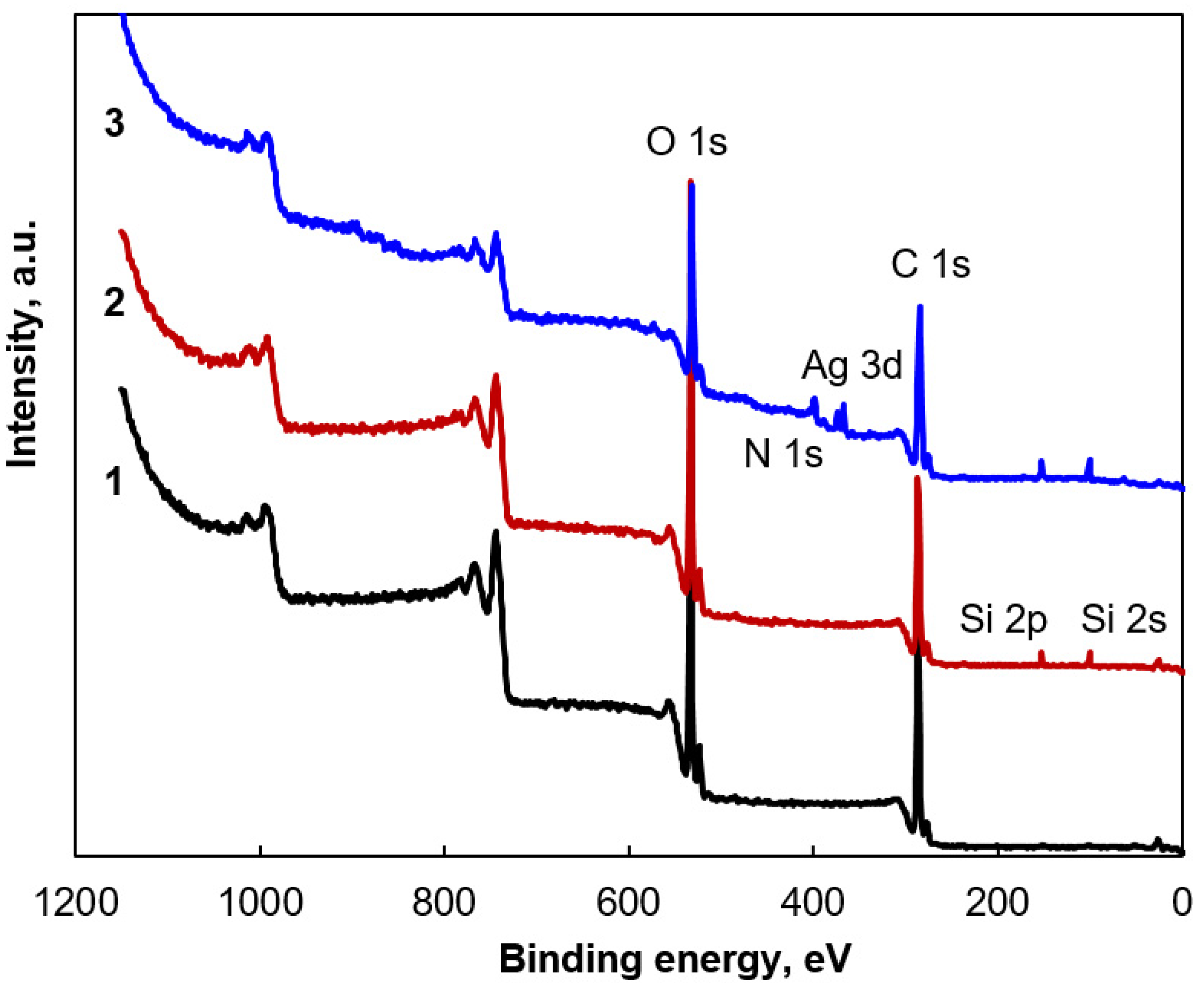
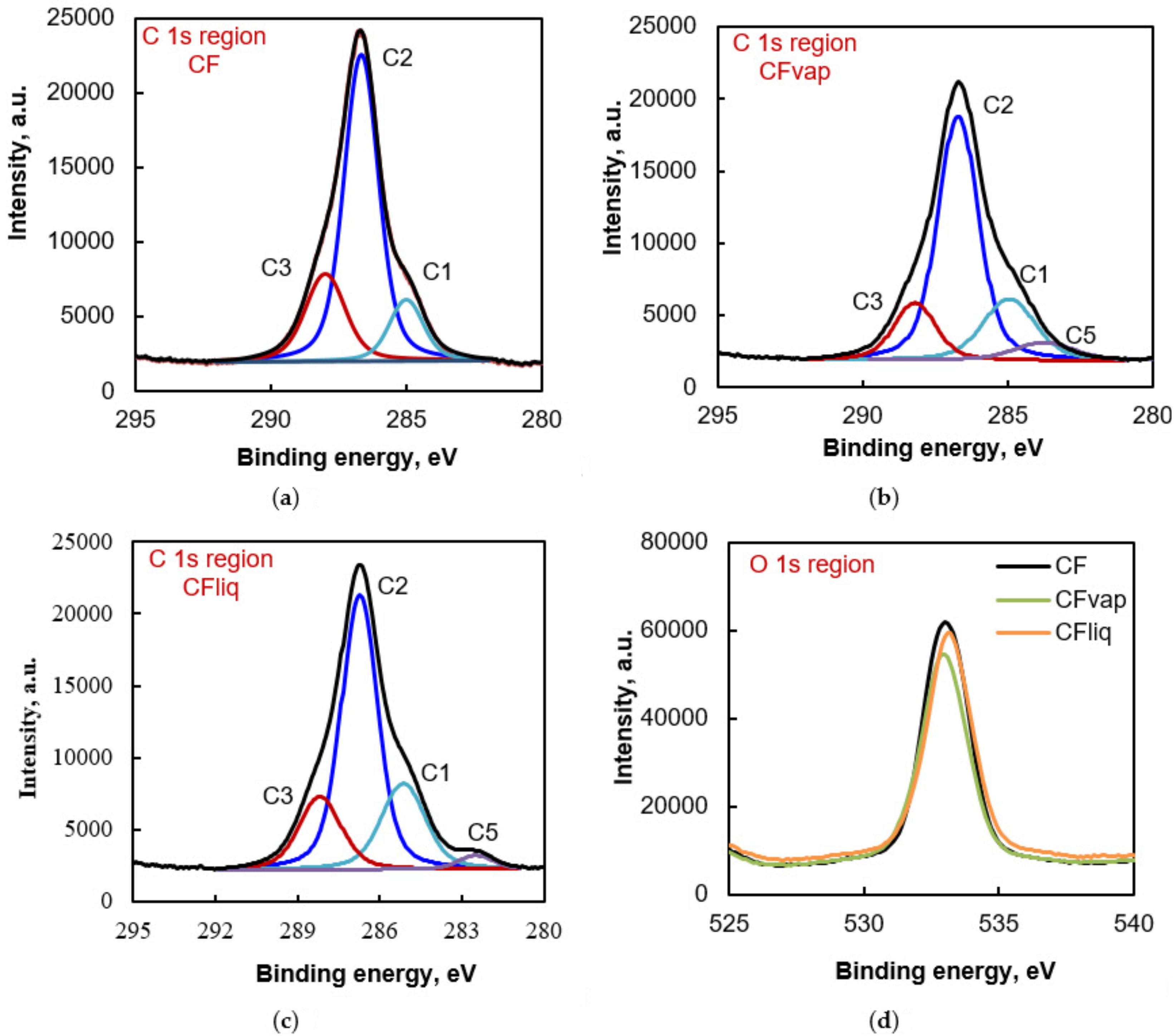
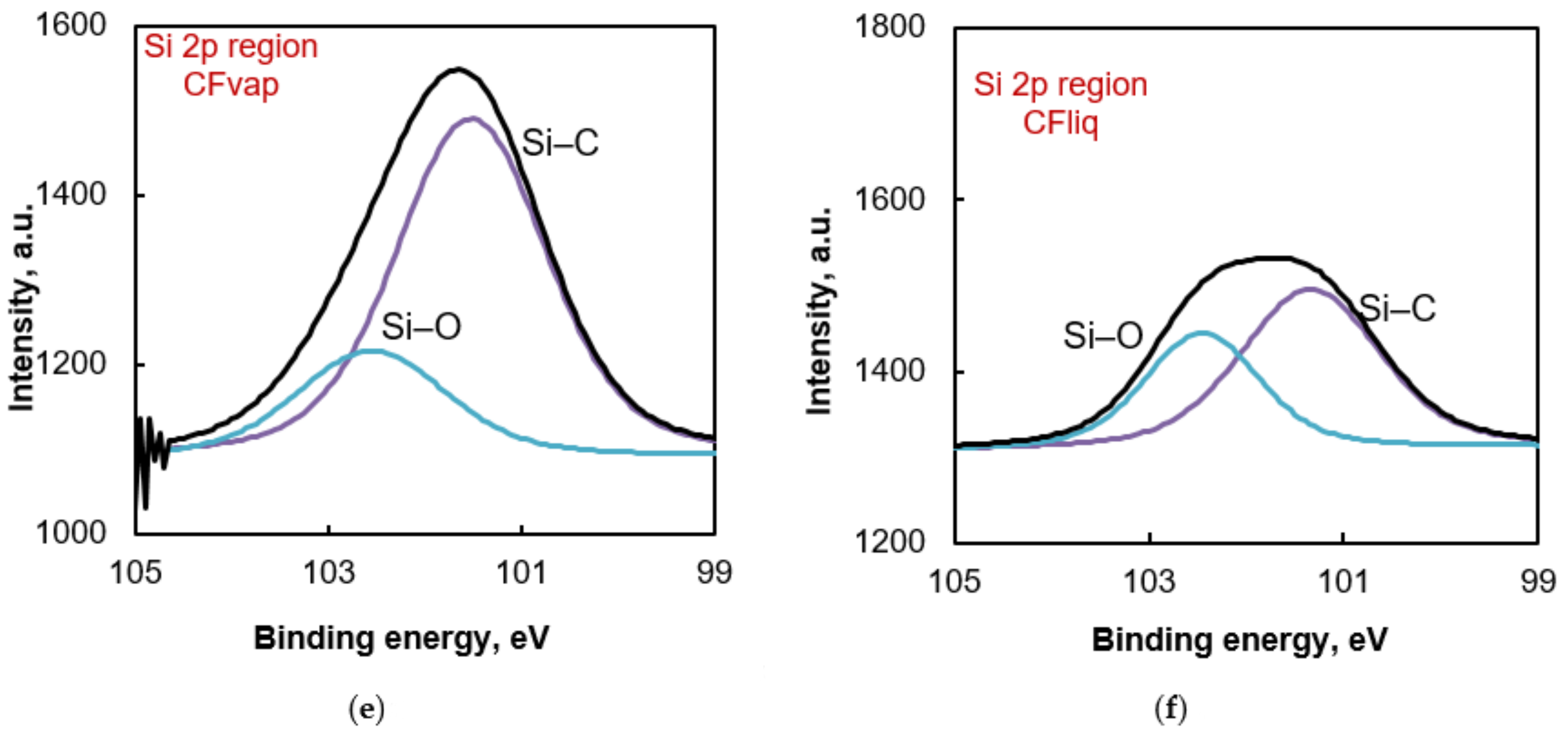
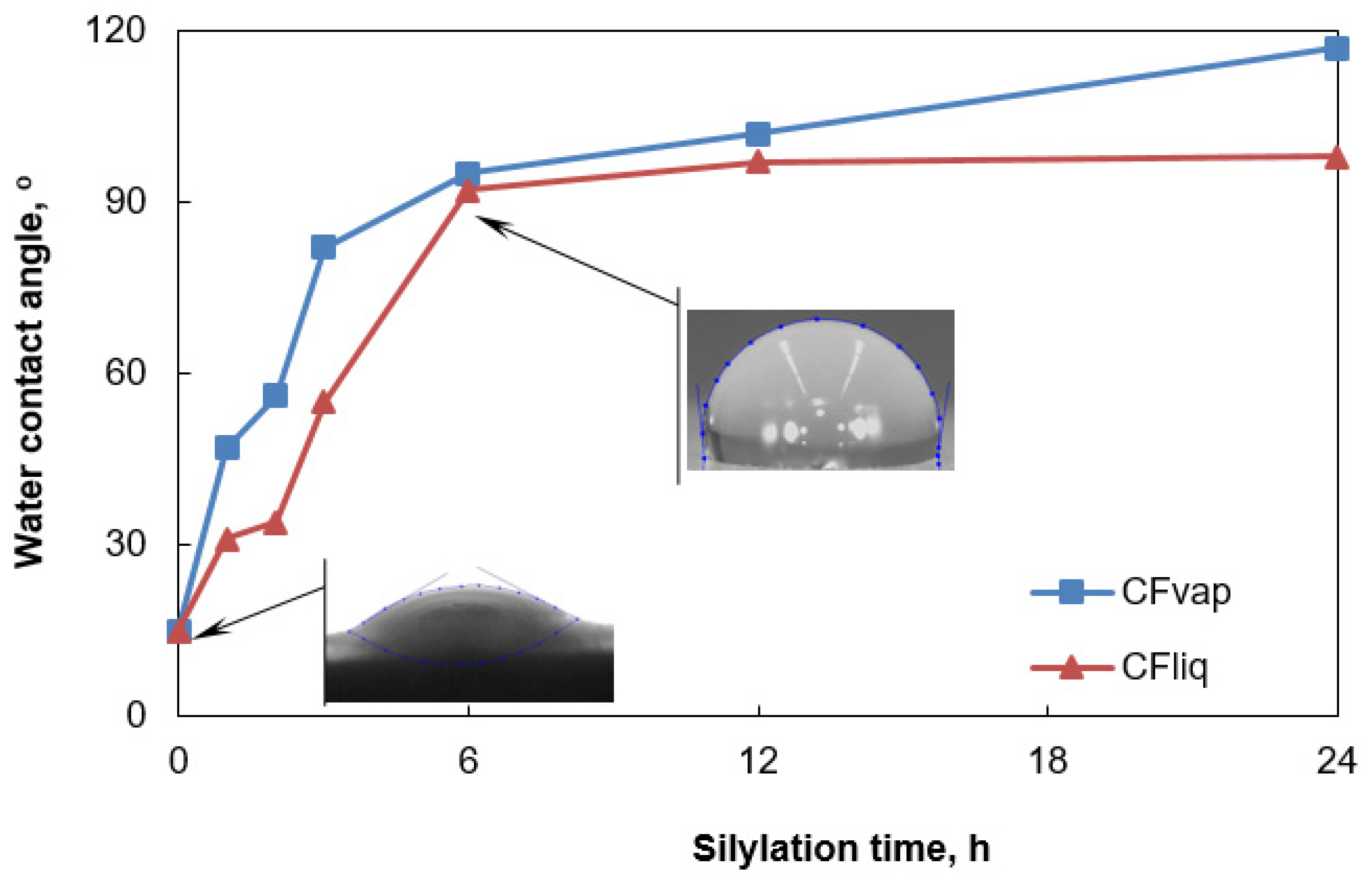
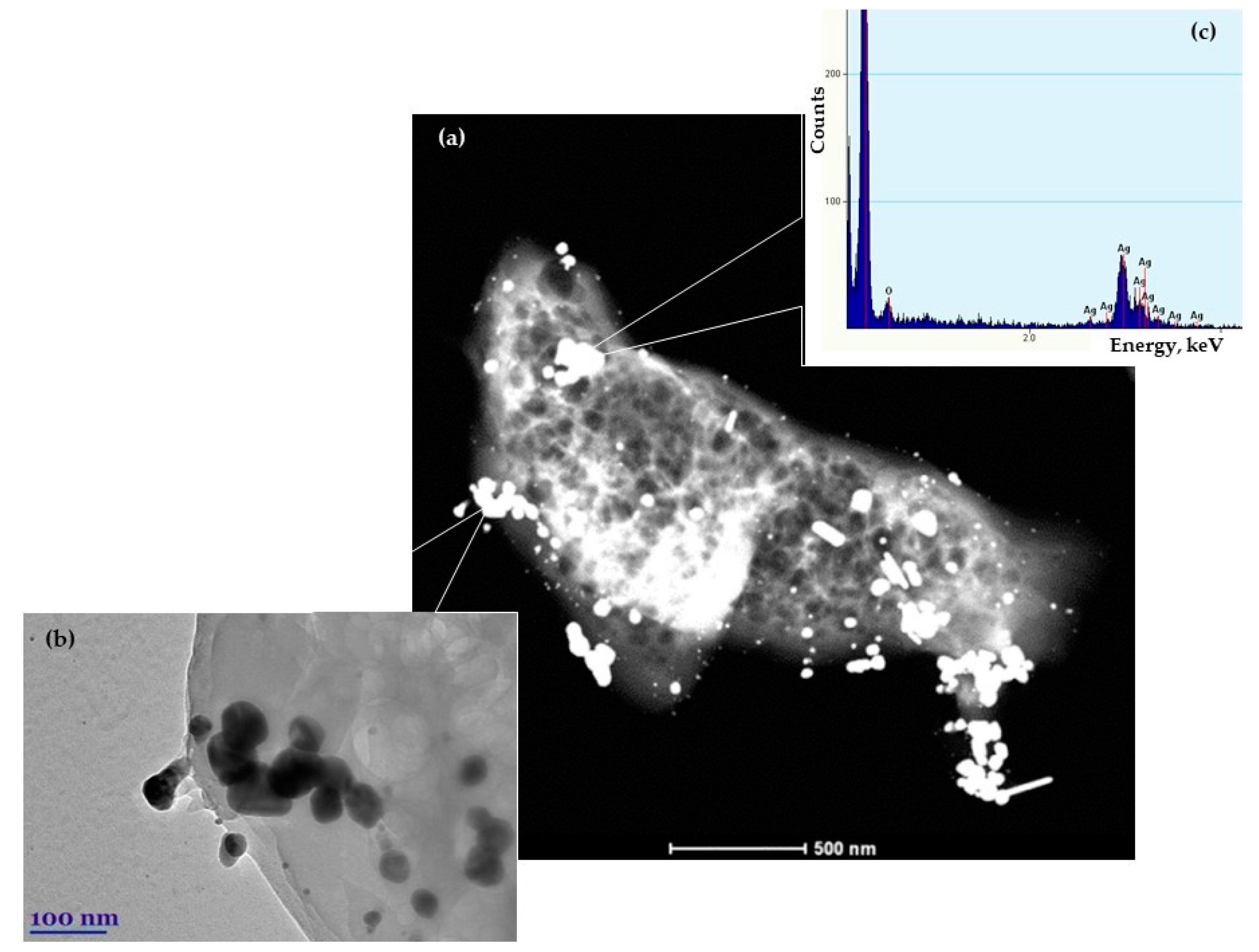
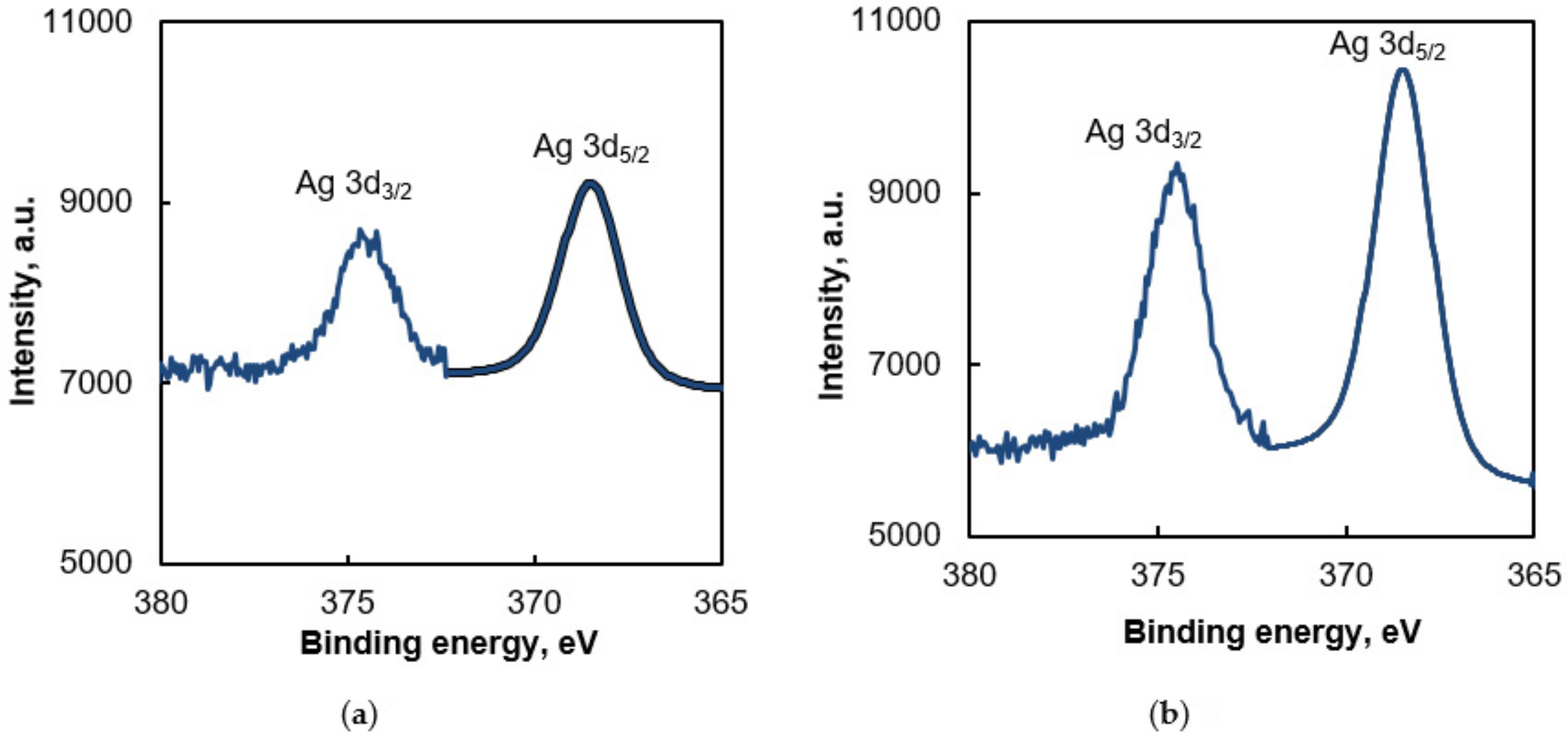
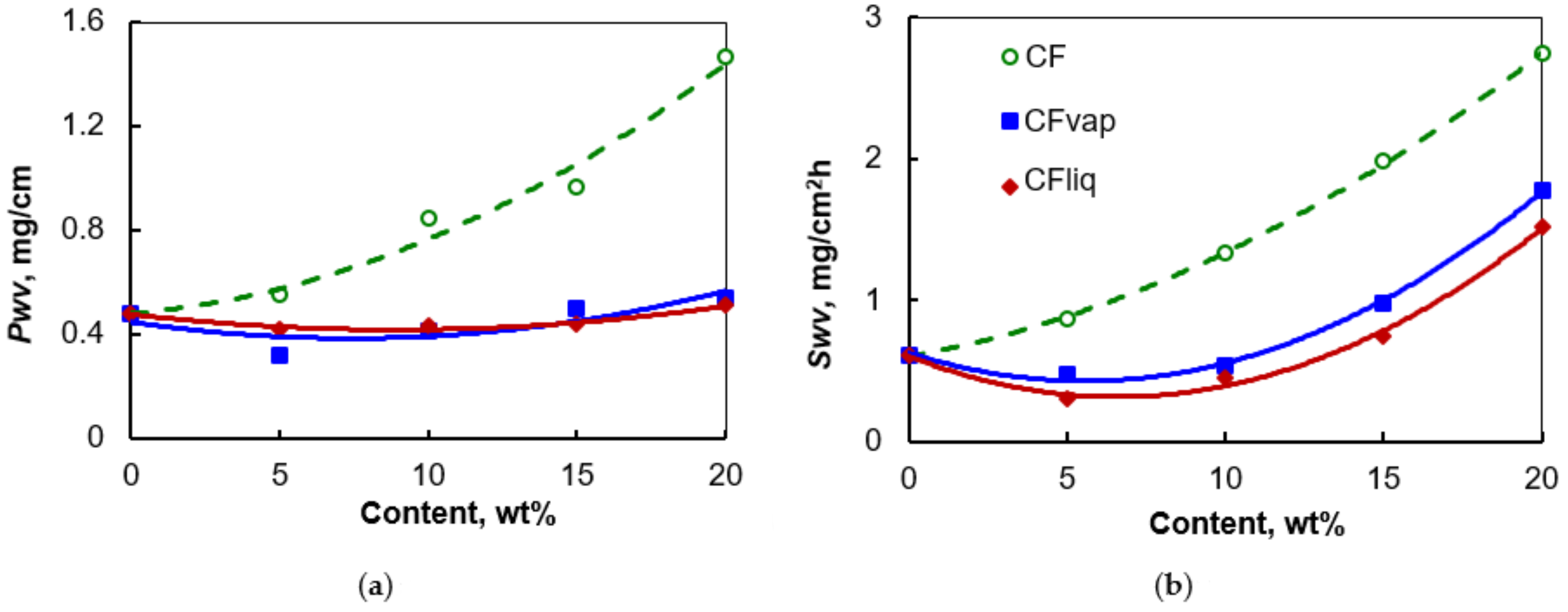
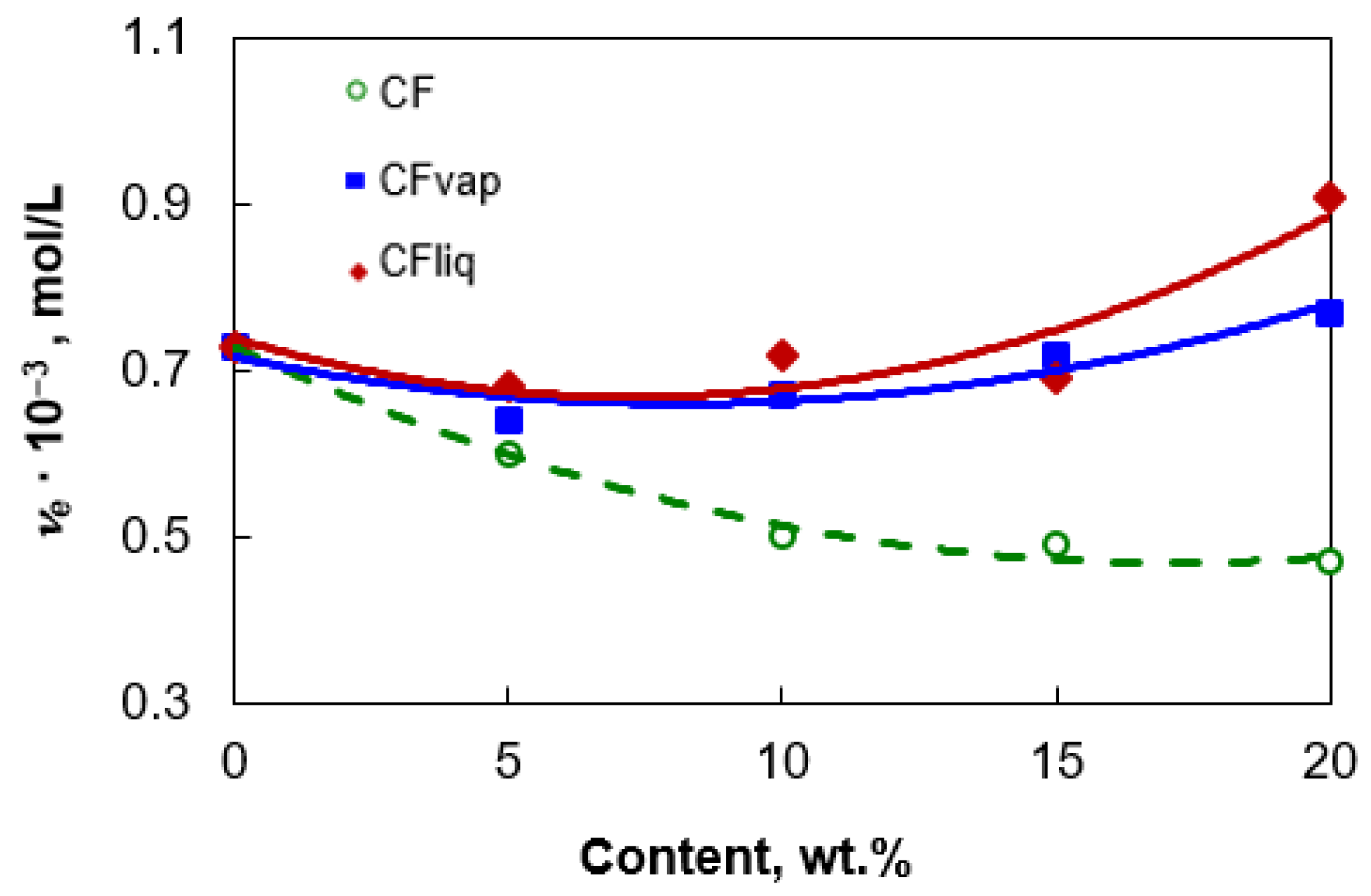
3.1. Morphology and Properties of Silylated CFs
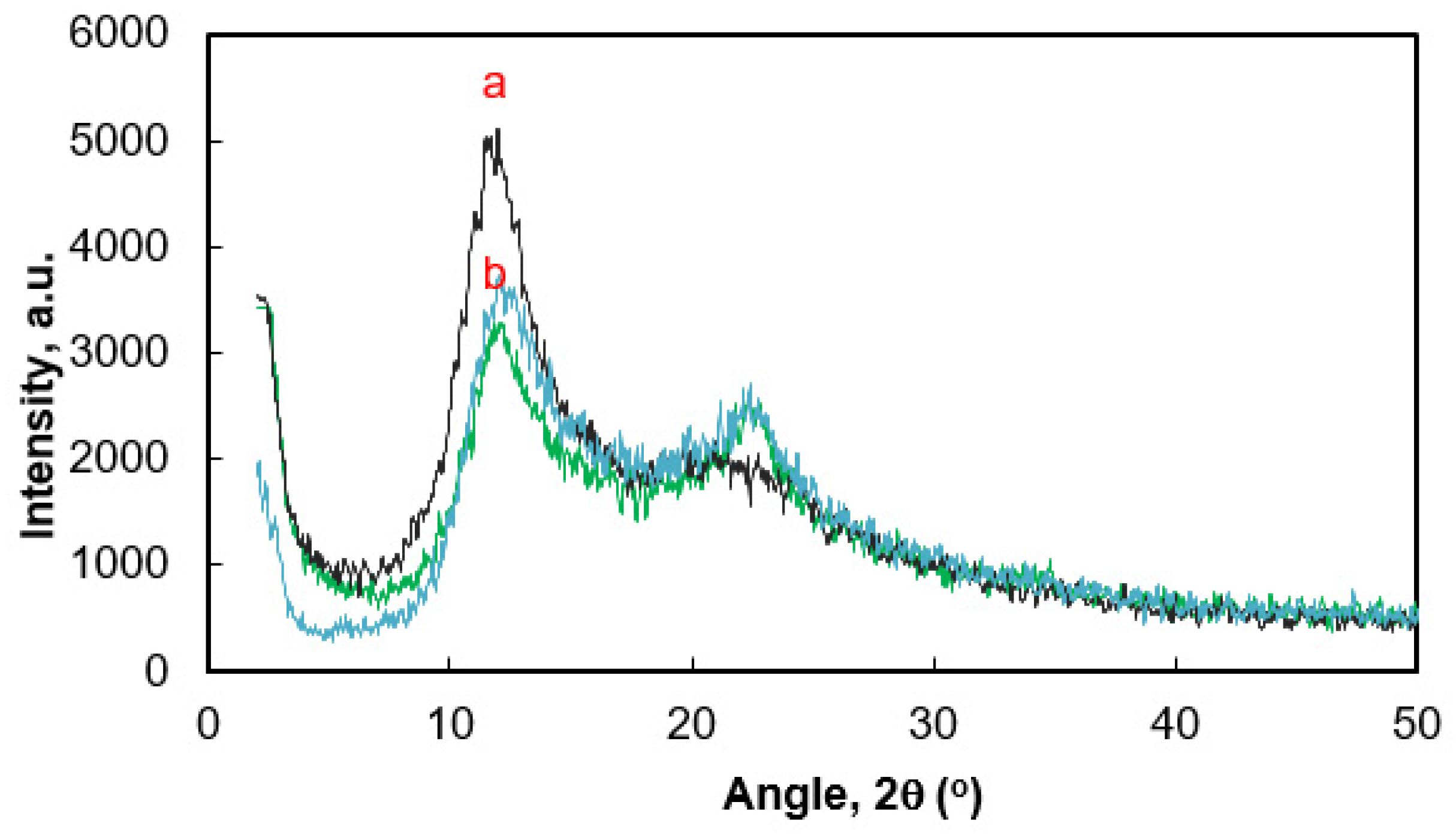
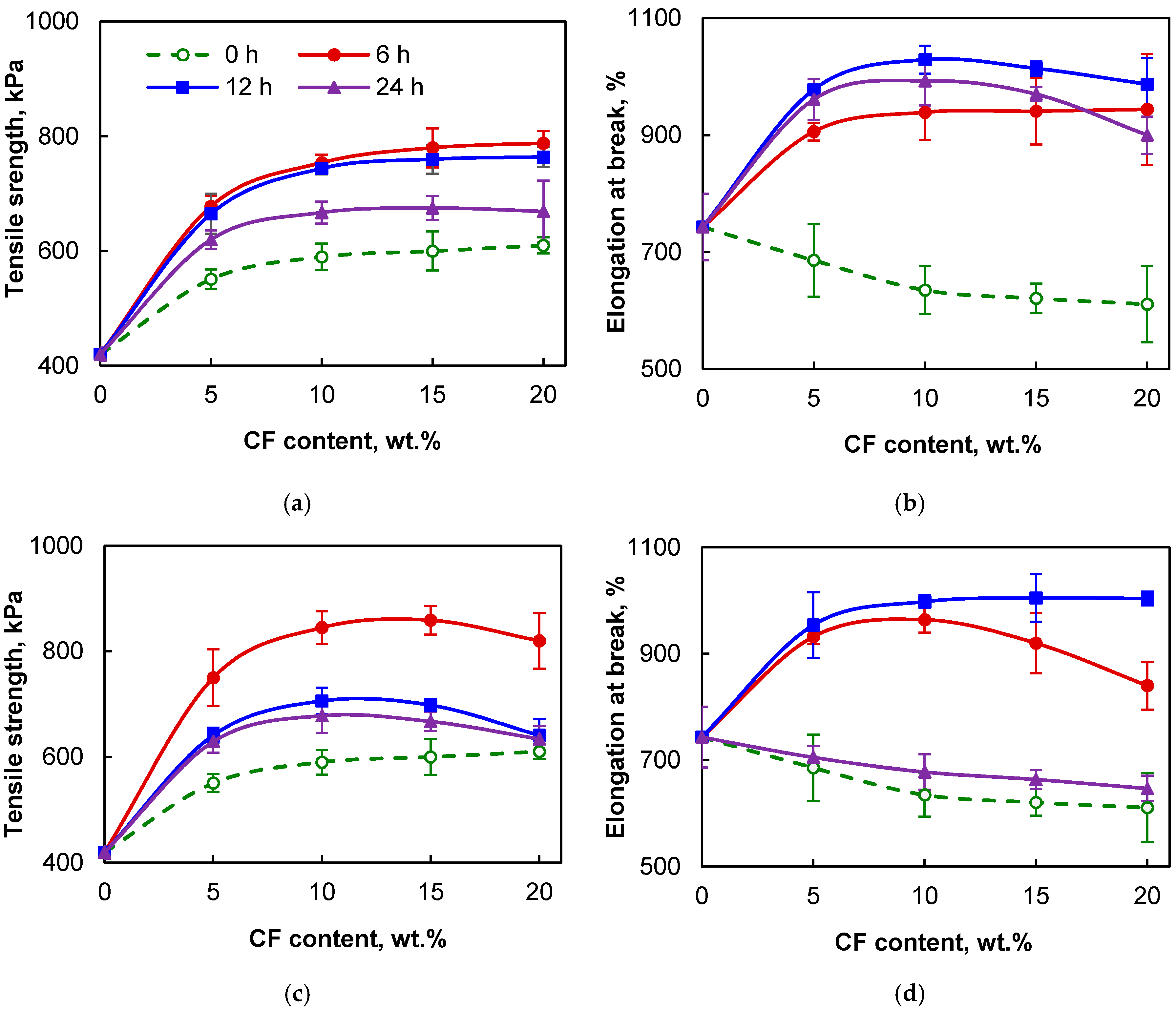
3.2. Structure and Properties of the PDMS/CF Composite
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ren, K.N.; Chen, Y.; Wu, H.K. New materials for microfluidics in biology. Curr. Opin. Biotechnol. 2014, 25, 78–85. [Google Scholar] [CrossRef]
- Gao, W.; Wang, X.; Xu, W. Magneto-mechanical properties of polydimethylsiloxane composites with a binary magnetic filler system. Polym. Composite 2019, 40, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Hwang, J.-Y.; Hwang, H.R. Simple and cost-effective method of highly conductive and elastic carbon nanotube/polydimethylsiloxane composite for wearable electronics. Sci. Rep. 2018, 8, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, J.; Colas, A. Medical applications of silicones. In Biomaterials Science: An Introduction to Materials in Medicine, 3rd ed.; Hoffman, B.D., Schoen, A.S., Lemons, F.J., Ratner, J.E., Eds.; Elsevier: Berlin, Germany, 2013; pp. 1106–1116. [Google Scholar]
- Kong, K.T.S.; Mariatti, M.; Rashid, A.A.; Busfield, J.J.C. Enhanced conductivity behavior of polydimethylsiloxane (PDMS) hybrid composites containing exfoliated graphite nanoplatelets and carbon nanotubes. Compos. B 2014, 58, 457–462. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Bai, S.; Yuan, X. Carbon fibre/graphene foam/polymer composites with enhanced mechanical and thermal properties. Compos. B Eng. 2016, 94, 102–108. [Google Scholar] [CrossRef]
- Smitha, A.; Rajeev, R.S.; Krishnaraj, K.; Sreenivas, N.; Manu, S.K.; Gouri, C.; Sekkar, V. Thermal protection characteristics of polydimethylsiloxane-organoclay nanocomposite. Polym. Degrad. Stab. 2017, 144, 281–291. [Google Scholar] [CrossRef]
- Alexandru, M.; Cazacu, M.; Vlad, S. Polydimethylsiloxane-silica composites. Influence of the silica on the morphology and the surface, thermal, mechanical properties. High Perform. Polym. 2009, 21, 379–392. [Google Scholar] [CrossRef]
- Gowda, Y.T.G.; Sanjay, M.R.; Bhat, K.S.; Madhu, P.; Senthamaraikannan, P.; Yogesha, B. Polymer matrix-natural fiber composites: An overview. Cogent. Eng. 2018, 5, 1446667. [Google Scholar]
- Istva, S.; Plackett, D. Microfibrillated cellulose and new nanocomposite materials: A review. Cellulose 2010, 17, 459–494. [Google Scholar]
- Panaitescu, D.M.; Frone, A.N.; Ghiurea, M.; Spataru, C.I.I.; Radovici, C.; Iorga, M.D. Properties of polymer composites with cellulose microfibrils. In Advances in Composite Materials—Ecodesign and Analysis; Attaf, B., Ed.; InTech: Nanterre, France, 2011; pp. 103–122. [Google Scholar]
- Palange, C.; Johns, M.A.; Scurr, D.J.; Phipps, J.S.; Eichhorn, S.J. The effect of the dispersion of microfibrillated cellulose on the mechanical properties of melt-compounded polypropylene–polyethylene copolymer. Cellulose 2019, 26, 9645–9659. [Google Scholar] [CrossRef] [Green Version]
- Jankauskaitė, V.; Abzalbekuly, B.; Lisauskaitė, A.; Procyčevas, I.; Fataraitė, E.; Vitkauskienė, A.; Janakhmetov, U. Silicone rubber and microcrystalline cellulose composites with antimicrobial properties. Mater. Sci. 2014, 20, 42–49. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Lu, A.; Zhang, L. Advances in cellulose hydrophobicity improvement. In Food Additives and Packaging; ACS: Washington, WA, USA, 2014; pp. 241–274. [Google Scholar]
- Valášek, P.; Müller, M.; Šleger, V. Influence of plasma treatment on mechanical properties of cellulose-based fibres and their interfacial interaction in composite systems. Bioresources 2017, 12, 5449–5461. [Google Scholar] [CrossRef]
- Belgacem, M.; Gandini, A. The surface modification of cellulose fibers for use as reinforcing elements in composite materials. Compos. Interface 2005, 12, 41–75. [Google Scholar] [CrossRef]
- Yeo, J.S.; Hwang, S.H. Preparation and characteristics of polypropylene-graft-maleic anhydride anchored micro-fibriled cellulose: Its composites with polypropylene. J. Adhes. Sci. Technol. 2015, 29, 185–194. [Google Scholar] [CrossRef]
- Jeon, J.G.; Kim, H.C.H.; Kim, J.; Kang, T.J. Polystyrene nanocomposites reinforced with phenyl isocyanate-treated cellulose nanofibers. Funct. Compos. Struct. 2020, 2, 015002. [Google Scholar] [CrossRef]
- Jeun, J.-P.; Lee, Y.; Kang, P.-H.; Lee, D.D.-Y. Surface modification for the hydrophobization of cellulose nanocrystals using radiation-induced grafting. J. Nanosci. Nanotechnol. 2019, 19, 6303–6308. [Google Scholar] [CrossRef]
- Boujemaoui, A.; Sanchez, C.C.; Engström, J.; Bruce, C.; Fogelström, L.; Carlmark, A.; Malmström, E. Polycaprolactone nanocomposites reinforced with cellulose nanocrystals surface-modified via covalent grafting or physisorption: A comparative study. ACS Appl. Mater. Interfaces 2017, 9, 35305–35318. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Franco, P.J.; Valadez-Gonzalez, A. A study of the mechanical properties of short natural-fiber reinforced composites. Compos. B 2005, 36, 597–608. [Google Scholar] [CrossRef]
- Abdelmouleh, M.; Boufi, S.; ben Salah, A.; Belgacem, M.N.; Gandini, A. Interaction of silane coupling agents with cellulose. Langmuir 2002, 18, 3203–3208. [Google Scholar] [CrossRef]
- Andresen, M.; Johansson, L.-S.; Tanem, B.S.; Stenius, P. Properties and characterization of hydrophobized microfibrillated cellulose. Cellulose 2006, 13, 665–677. [Google Scholar] [CrossRef]
- Gousse, C.; Chanzy, H.; Cerrada, M.L.; Fleury, E. Surface silylation of cellulose microfibrils: Preparation and rheological properties. Polymer 2004, 45, 1569–1575. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Chen, R.; Chen, G.-Y.; Liu, L.; Yang, X.-G.; Yao, J.-M. Silylation of cellulose nanocrystals and their reinforcement of commercial silicone rubber. J. Nanopart Res. 2015, 17, 361. [Google Scholar] [CrossRef]
- Chinga-Carrasco, G.; Kuznetsova, N.; Garaeva, M.; Leirset, I.; Galiullina, G.; Kostochko, A.; Syverud, K. Bleached and unbleached MFC nanobarriers: Properties and hydrophobisation with hexamethyldisilazane. J. Nanopar. Res. 2012, 14, 1280. [Google Scholar] [CrossRef]
- Mormann, W. Silylation of cellulose with hexamethyldisilazane in ammonia—Activation, catalysis, mechanism, properties. Cellulose 2003, 10, 271–281. [Google Scholar] [CrossRef]
- Jankauskaitė, V.; Narmontas, P.; Lazauskas, A. Control of polydimethylsiloxane surface hydrophobicity by plasma polymerized hexamethyldisilazane deposition. Coatings 2019, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Grunert, M.; Winter, W. Nanocomposites of cellulose acetate butyrate reinforced with cellulose nanocrystals. J. Polym. Environ. 2002, 10, 27–30. [Google Scholar] [CrossRef]
- Zhang, Z.; Tingaut, P.; Rentsch, D.; Zimmermann, T.; Sèbe, G. Controlled silylation of nanofibrillated cellulose in water: Reinforcement of a model polydimethylsiloxane network. ChemSusChem 2015, 8, 2681–2690. [Google Scholar] [CrossRef]
- Soliveri, G.; Meroni, D.; Cappelletti, G.; Annunziata, R.; Aina, V.; Cerrato, G.; Ardizzone, S. Engineered organic/inorganic hybrids for superhydrophobic coatings by wet and vapour procedures. J. Mater. Sci. 2014, 49, 2734–2744. [Google Scholar] [CrossRef]
- Zhang, F.; Sautter, K.; Larsen, A.M.; Findley, D.A.; Davis, R.C.; Samha, H.; Linford, M.R. Chemical vapour deposition of three aminosilanes on silicon dioxide: Surface characterization, stability, effects of silane concentration, and cyanine dye adsorption. Langmuir 2010, 26, 14648–14654. [Google Scholar] [CrossRef]
- Deng, S.; Binauld, S.; Mangiante, G.; Frances, J.M.; Charlot, A.; Bernard, J.; Zhou, X.; Fleury, E. Microcrystalline cellulose as reinforcing agent in silicone elastomers. Carbohyd. Polym. 2016, 151, 899–906. [Google Scholar] [CrossRef]
- Jankauskaitė, V.; Lazauskas, A.; Griškonis, E.; Lisauskaitė, A.; Žukienė, K. UV-curable aliphatic silicone acrylate organic-inorganic hybrid coatings with antibacterial activity. Molecules 2017, 22, e964. [Google Scholar] [CrossRef] [Green Version]
- Kottmann, A.; Mejía, E.; Hémery, T.; Klein, J.; Kragl, U. Recent developments on the preparation of silicones with antimicrobial properties. Chem. Asian J. 2017, 12, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Chen, H. Mechanical and antibacterial properties of silicone rubber compounds filled with silver-zirconium phosphate modified by aluminate coupling agent. Polym. Plast. Technol. 2017, 56, 1969–1976. [Google Scholar] [CrossRef]
- Singh, R.; Shedbalkar, U.U.; Wadhwani, S.A.; Chopade, B.A. Bacteriagenic silver nanoparticles: Synthesis, mechanism, and applications. Appl. Microbiol. Biotechnol. 2015, 99, 4579–4593. [Google Scholar] [CrossRef]
- Durán, N.; Durán, M.; de Jesus, M.B.; Seabra, A.B.; Fávaro, W.J.; Nakazato, G. Silver nanoparticles: A new view on mechanistic aspects on antimicrobial activity. Nanomed. Nanotechnol. 2016, 12, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Greulich, C.; Braun, D.; Peetsch, A.; Diendorf, J.; Siebers, B.; Epple, M.; Köller, M. The toxic effect of silver ions and silver nanoparticles towards bacteria and human cells occurs in the same concentration range. RSC Adv. 2012, 2, 6981–6987. [Google Scholar] [CrossRef]
- Ferdous, Z.; Nemmar, A. Health Impact of Silver Nanoparticles: A review of the biodistribution and toxicity following various routes of exposure. Int. J. Mol. Sci. 2020, 21, 2375. [Google Scholar] [CrossRef] [Green Version]
- Groza, A.; Ciobanu, C.S.; Popa, C.L.; Iconaru, S.L.; Chapon, P.; Luculescu, C.; Predoi, D. Tructural properties and antifungal activity against candida albicans biofilm of different composite layers based on Ag/Zn doped hydroxyapatite-polydimethylsiloxanes. Polymers 2016, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- de Moraes, A.C.M.; Lima, B.A.; de Faria, A.F.; Brocchi, M.; Alves, O.L. Graphene oxide-silver nanocomposite as a promising biocidal agent against methicillin-resistant Staphylococcus aureus. Int. J. Nanomed. 2015, 10, 6847–6861. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, S.; Yue, X.; Lu, W. Review of silver nanoparticles (AgNPs)-cellulose antibacterial composites. Bioresources 2018, 13, 2150–2170. [Google Scholar] [CrossRef]
- Dong, Y.-Y.; Fu, L.-H.; Liu, S.; Ma, M.-G.; Wang, B. Silver-reinforced cellulose hybrids with enhanced antibacterial activity: Synthesis, characterization, and mechanism. RSC Adv. 2015, 5, 97359–97366. [Google Scholar] [CrossRef]
- Chen, G.; Yan, L.; Wan, X.; Zhang, Q.; Wang, Q. In situ synthesis of silver nanoparticles on cellulose fibers using D-glucuronic acid and its antibacterial application. Materials 2019, 12, 3101. [Google Scholar] [CrossRef] [Green Version]
- Bronstein, L.M.; Shifrina, S.B. Dendrimers as encapsulating, stabilizing, or directing agents for inorganic nanoparticles. Chem. Rev. 2011, 111, 5301–5344. [Google Scholar] [CrossRef] [PubMed]
- Balčiūnaitienė, A. Development and Investigation of Antimicrobial Polydimethylsiloxane Composites. Ph.D. Thesis, Kaunas University of Technology, Kaunas, Lithuania, 14 February 2019. [Google Scholar]
- Visakh, P.M.; Thomas, S. Preparation of bionanomaterials and their polymer nanocomposites from waste and biomass. Waste Biomass Valorization 2010, 1, 121–134. [Google Scholar] [CrossRef]
- Fu, P.F.; Glover, S.; King, R.K.; Lee, C.I.; Pretzer, M.R.; Tomalia, M.K. Polypropylene–polysiloxane block copolymers via hydrosilylation of monovinylidene capped isotactic polypropylene. ACS Polym. Prepr. 2003, 44, 1014–1015. [Google Scholar]
- Stalder, A.; Kulik, G.; Sage, D.; Barbieri, L.; Hoffmann, P. A snake-based approach to accurate determination of both contact points and contact angles. Colloid Surf. A 2006, 286, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J.; Rehner, J.J. Statistical mechanics of crosslinked polymer networks I. Rubber-like elasticity. J. Chem. Phys. 1943, 11, 512–520. [Google Scholar] [CrossRef]
- Poletto, M.; Pistor, V.; Zattera, A.J. Structural characteristics and thermal properties of native cellulose. In Cellulose—Fundamental Aspects; IntechOpen: London, UK, 2013; pp. 45–68. [Google Scholar]
- Hospodarova, V.; Singovszka, E.; Stevulova, N. Characterization of cellulosic fibers by ftir spectroscopy for their further implementation to building materials. Am. J. Anal. Chem. 2018, 9, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Popescua, M.C.; Popescua, C.-M.; Lisab, G.; Sakatac, Y. Evaluation of morphological and chemical aspects of different wood species by spectroscopy and thermal methods. J. Mol. Struct. 2011, 988, 65–72. [Google Scholar] [CrossRef]
- Jankowska, I.; Ławniczak, P.; Pogorzelec-Glaser, K.; Łapinski, A.; Pankiewicz, R.; Tritt-Goc, J. Cellulose microfibers surface treated with imidazole as new proton conductors. J. Mater. Chem. Phys. 2020, 239, 122056. [Google Scholar] [CrossRef]
- Carrilo, F.; Colom, X.; Sunol, J.J.; Saurina, J. Structural FTIR analysis and thermal characterization of lyocell and viscose-type fibres. Eur. Polym. J. 2004, 40, 2229–2234. [Google Scholar] [CrossRef]
- Åkerholm, M.; Hinterstoisser, B.; Salmén, L. Characterization of the crystalline structure of cellulose using static and dynamic FT-IR spectroscopy. Carbohydr. Res. 2004, 339, 569–578. [Google Scholar] [CrossRef]
- Xie, B.; Muscat, A.J. Silylation of porous methylsilsesquioxane film in suoercritical carbon dioxide. Microelect. Eng. 2004, 76, 52–59. [Google Scholar] [CrossRef]
- Che, M.-L.; Teng, J.-Y.; Lai, P.-C.; Leu, J. Moisture uptake and dielectric property of methylsilsesquioxane/high-temperature porogen hybrids and porous low-k films. J. Mater. Res. 2011, 26, 2987–2995. [Google Scholar] [CrossRef] [Green Version]
- Souguir, Z.; Dupont, A.-L.; Fatyeyeva, K.; Mortha, G.; Cheradame, H.; Ipert, S.; Lavédrine, B. Strengthening of degraded cellulosic material using a diamine alkylalkoxysilane. RSC Adv. 2012, 2, 7470–7478. [Google Scholar] [CrossRef]
- Kim, H.; Youn, J.R.; Song, Y.S. Eco-friendly flame retardant nanocrystalline cellulose prepared via silylation. Nanotechnology 2018, 29, 455702. [Google Scholar] [CrossRef]
- Liu, L.; Xie, J.P.; Li, Y.J.; Zhang, Q.; Yao, J. Three-dimensional macroporous cellulose-based bioadsorbents for efficient removal of nickel ions from aqueous solution. Cellulose 2015, 23, 723–736. [Google Scholar] [CrossRef]
- Samana, N.; Johari, K.; Kongc, H.; Mohtar, S.S.; Hassana, O.; Ali, N.; Mat, H. Enhanced elemental mercury removal by facile sulfurization of agrowaste chars. Chem. Eng. Res. Des. 2019, 144, 198–208. [Google Scholar] [CrossRef]
- Frone, A.N.; Panaitescu, D.M.; Chiulan, I.; Nicolae, C.-A.; Casarica, A.; Nicolae, C.-A.; Trusca, R.; Damian, C.; Purcar, V.; Alexandrescu, E.; et al. Surface treatment of bacterial cellulose in mild, eco-friendly conditions. Coatings 2018, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.-J.; Wang, M.; Zhang, X.-Y.; Wu, J.-Y.; Zhang, T. Investigation on the role of the molecular weight of polyvinyl pyrrolidone in the shape control of high-yield silver nanospheres and nanowires. Nanoscale Res. Lett. 2014, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozovskis, P.; Jankauskaitė, V.; Guobienė, A.; Kareivienė, V.; Vitkauskienė, A. Effect of graphene oxide and silver nanoparticles hybrid composite on P. aeruginosa strains with acquired resistance genes. Int. J. Nanomed. 2020, in press. [Google Scholar]
- Ferraria, A.M.; Boufi, S.; Battaglini, N.; Botelho do Rego, A.M.; ReiVilar, M. Hybrid systems of silver nanoparticles generated on cellulose surfaces. Langmuir 2010, 26, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Han, S.W.; Kim, Y.; Kim, K. Dodecanethiol-derivatized Au/Ag bimetallic nanoparticles: TEM, UV/VIS, XPS, and FTIR analysis. J. Colloid. Interface Sci. 1998, 208, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, A.; Haddadi, A.A.; Mahdavian, M. The effect of sol–gel surface modified silver nanoparticles on the protective properties of the epoxy coating. RSC Adv. 2016, 6, 18996–19006. [Google Scholar] [CrossRef]
- Barud, H.S.; Regiani, T.; Marques, R.F.C.; Lustri, W.R.; Messaddeq, Y.; Ribeiro, S.J. Antimicrobial bacterial cellulose-silver nanoparticles composite membranes. J. Nanomater. 2011, 2011, 721631. [Google Scholar] [CrossRef] [Green Version]
- Morones, J.R.; Elechiguerra, J.L.; Camacho, A.; Morones, J.R.; Elechiguerra, J.L.; Camacho, A.; Holt, K.; Kouri, J.B.; Ramírez, J.T.; Yacaman, M.J. The bactericidal effect of silver nanoparticles. Nanotechnology 2005, 16, 2346–2353. [Google Scholar] [CrossRef] [Green Version]
- Mangiante, G.; Alcouffe, P.; Burdin, B.; Gaborieau, M.; Zeno, E.; Petit-Conil, M.; Bernard, J.; Charlot, A.; Fleury, E. Green nondegrading approach to alkyne-functionalized cellulose fibers and biohybrids thereof: Synthesis and mapping of the derivatization. Biomacromolecules 2013, 14, 254–263. [Google Scholar] [CrossRef]
- Fröhlich, J.; Niedermeier, W.; Luginsland, H.-D. The effect of filler–filler and filler–elastomer interaction on rubber reinforcement. Compos. A Appl. Sci. Manuf. 2002, 36, 449–460. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankauskaitė, V.; Balčiūnaitienė, A.; Alexandrova, R.; Buškuvienė, N.; Žukienė, K. Effect of Cellulose Microfiber Silylation Procedures on the Properties and Antibacterial Activity of Polydimethylsiloxane. Coatings 2020, 10, 567. https://doi.org/10.3390/coatings10060567
Jankauskaitė V, Balčiūnaitienė A, Alexandrova R, Buškuvienė N, Žukienė K. Effect of Cellulose Microfiber Silylation Procedures on the Properties and Antibacterial Activity of Polydimethylsiloxane. Coatings. 2020; 10(6):567. https://doi.org/10.3390/coatings10060567
Chicago/Turabian StyleJankauskaitė, Virginija, Aistė Balčiūnaitienė, Radostina Alexandrova, Nijolė Buškuvienė, and Kristina Žukienė. 2020. "Effect of Cellulose Microfiber Silylation Procedures on the Properties and Antibacterial Activity of Polydimethylsiloxane" Coatings 10, no. 6: 567. https://doi.org/10.3390/coatings10060567