Structural Basis of CO2 Adsorption in a Flexible Metal-Organic Framework Material

 , , ,
, , ,
Abstract
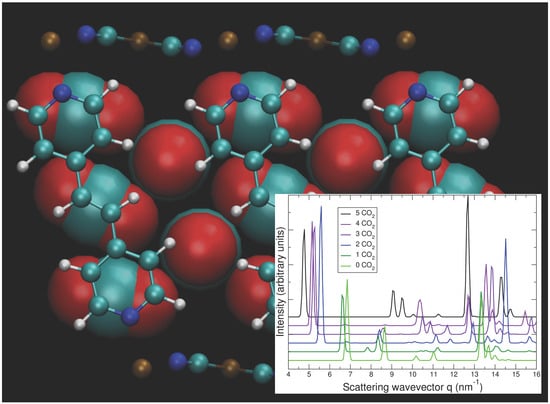
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Preparation of NiBpene Powder
2.2. Small-Angle Neutron Scattering and Diffraction under Static Dual Gas Conditions
2.3. Small-Angle X-Ray Scattering and Diffraction under Dual Gas Flow Conditions
2.4. Small-Angle X-Ray Scattering and Diffraction under Supercritical CO2 Conditions
2.5. Estimation of Lattice Parameter Changes in Response to Gas Adsorption/Desorption in NiBpene
2.6. DFT Model Interpretation
3. Results and Discussion
3.1. Dual Gas Adsorption under Static Pressure Conditions
3.2. Dual Gas Adsorption and Desorption under Flow Pressure Conditions
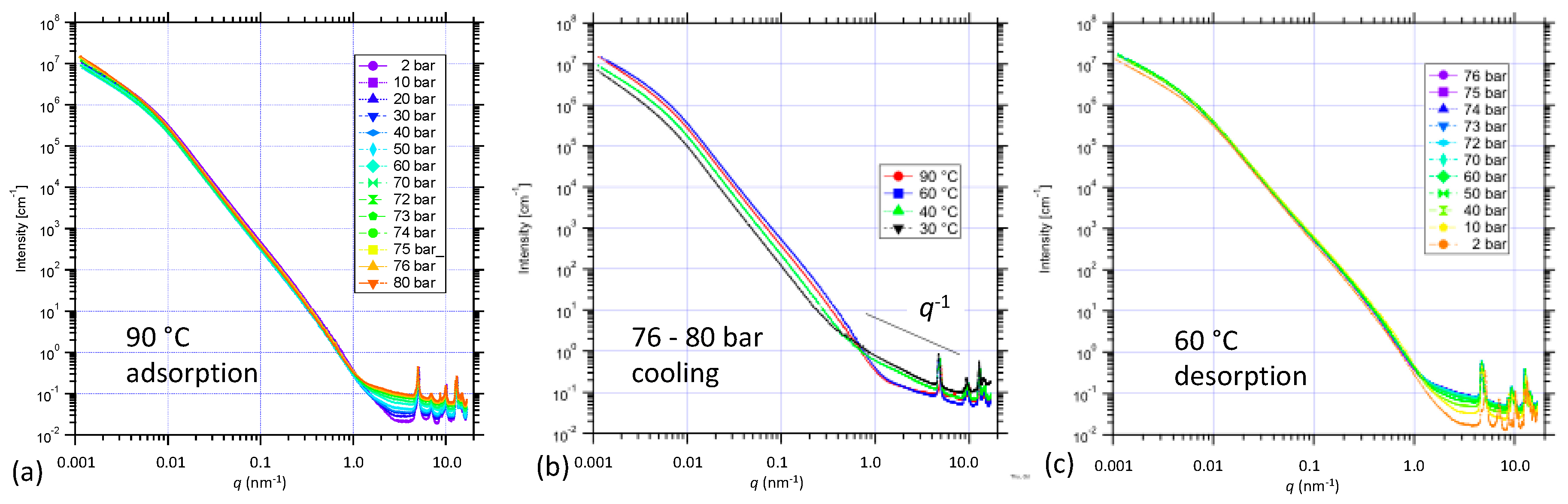
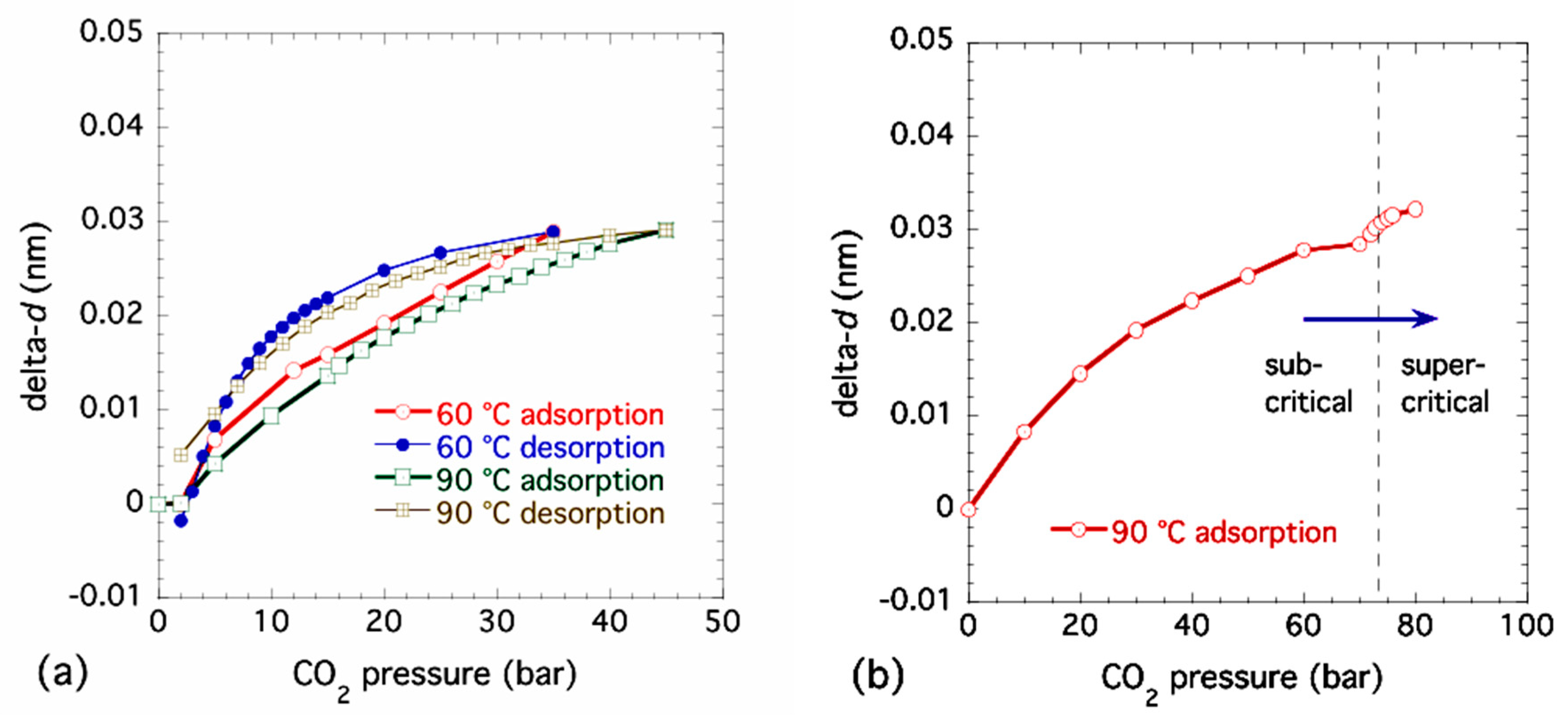
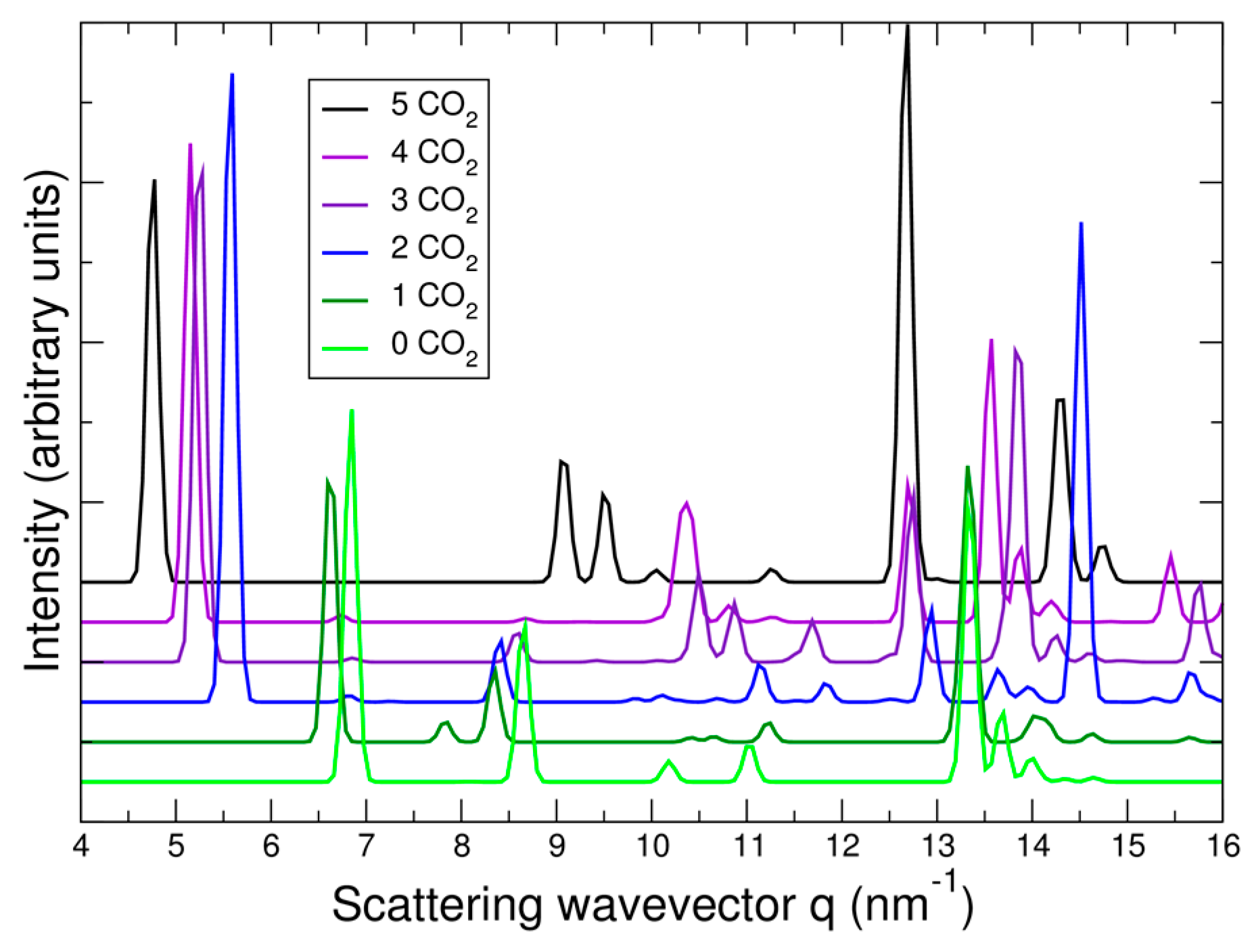
3.3. Structural Changes in NiBpene During CO2 Adsorption/Desorption under Supercritical Conditions
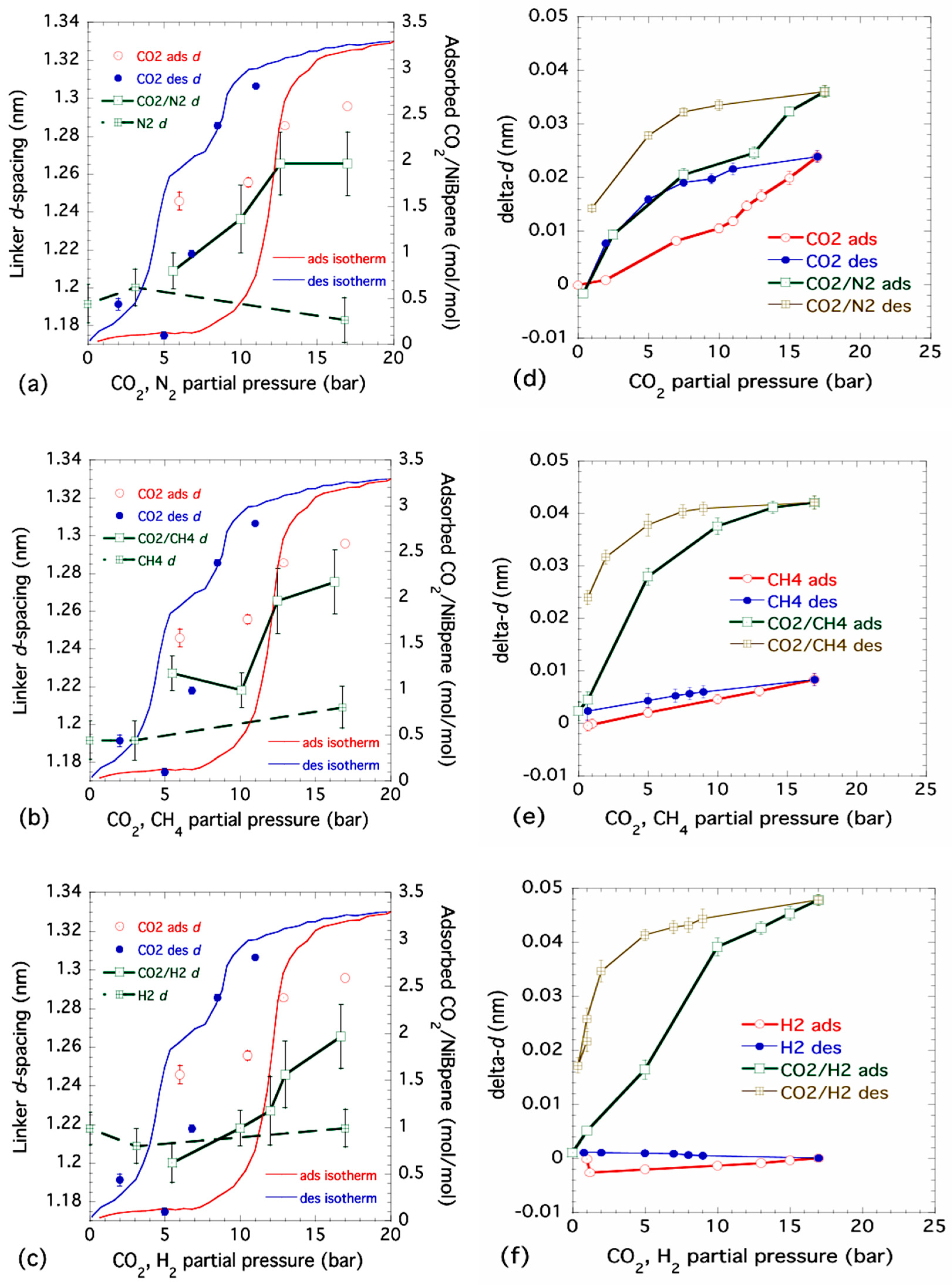
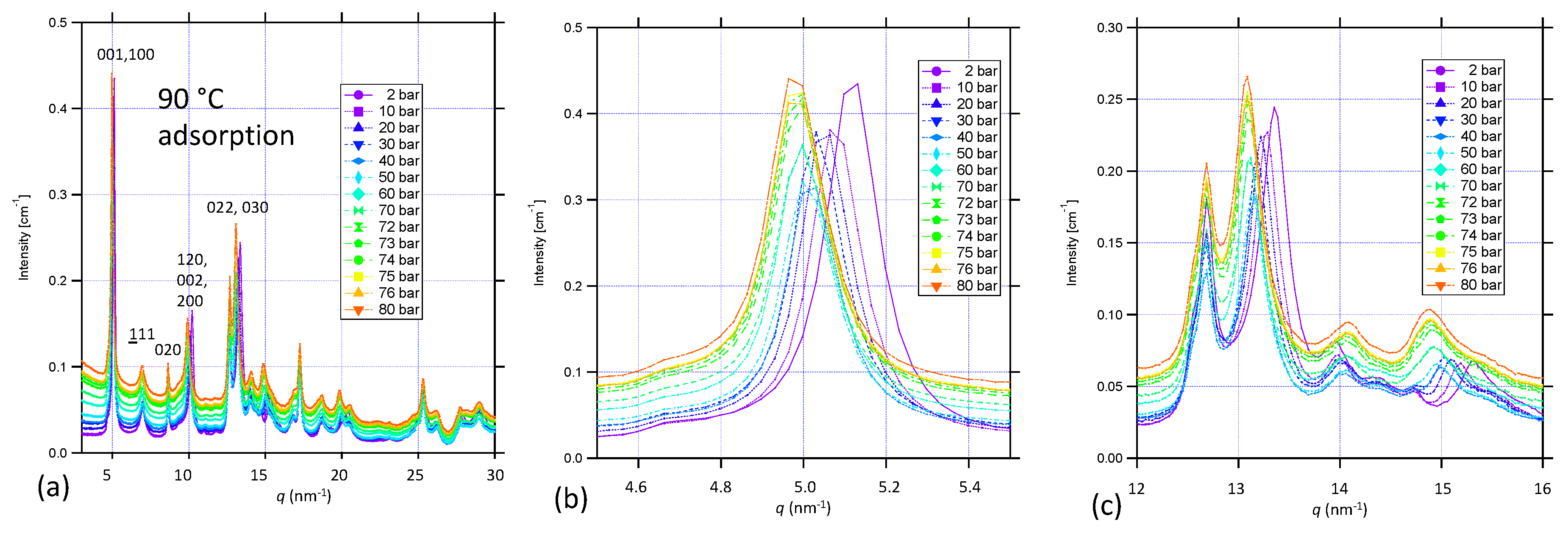
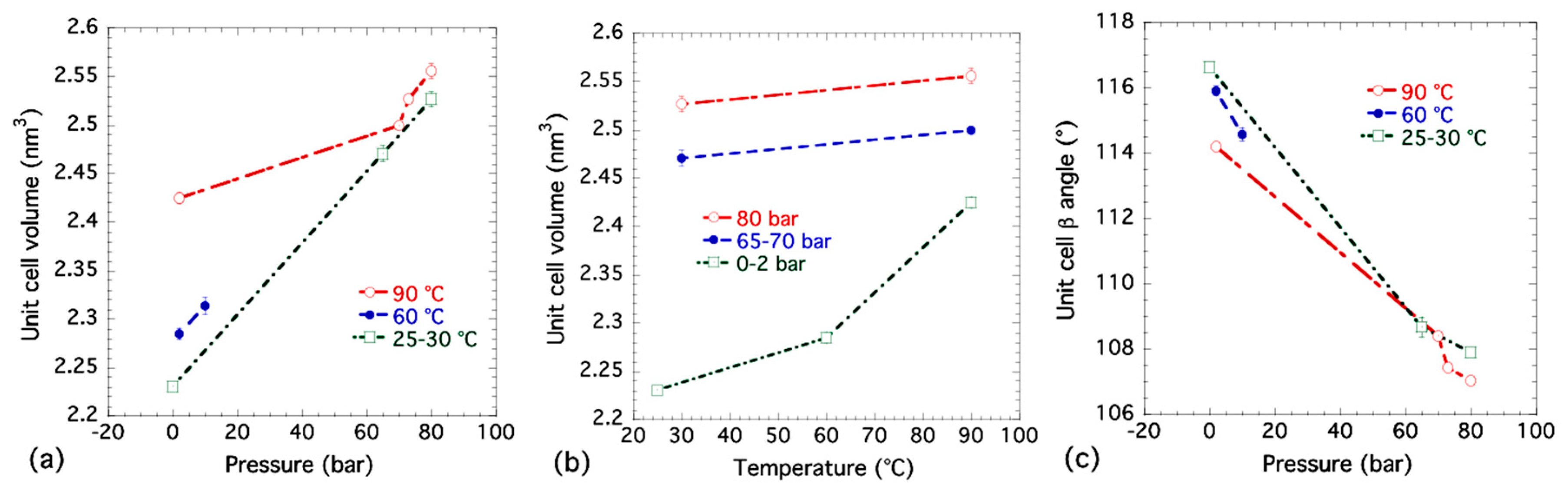
3.4. Lattice Parameter Determination of NiBpene under Selected CO2 Pressure Conditions
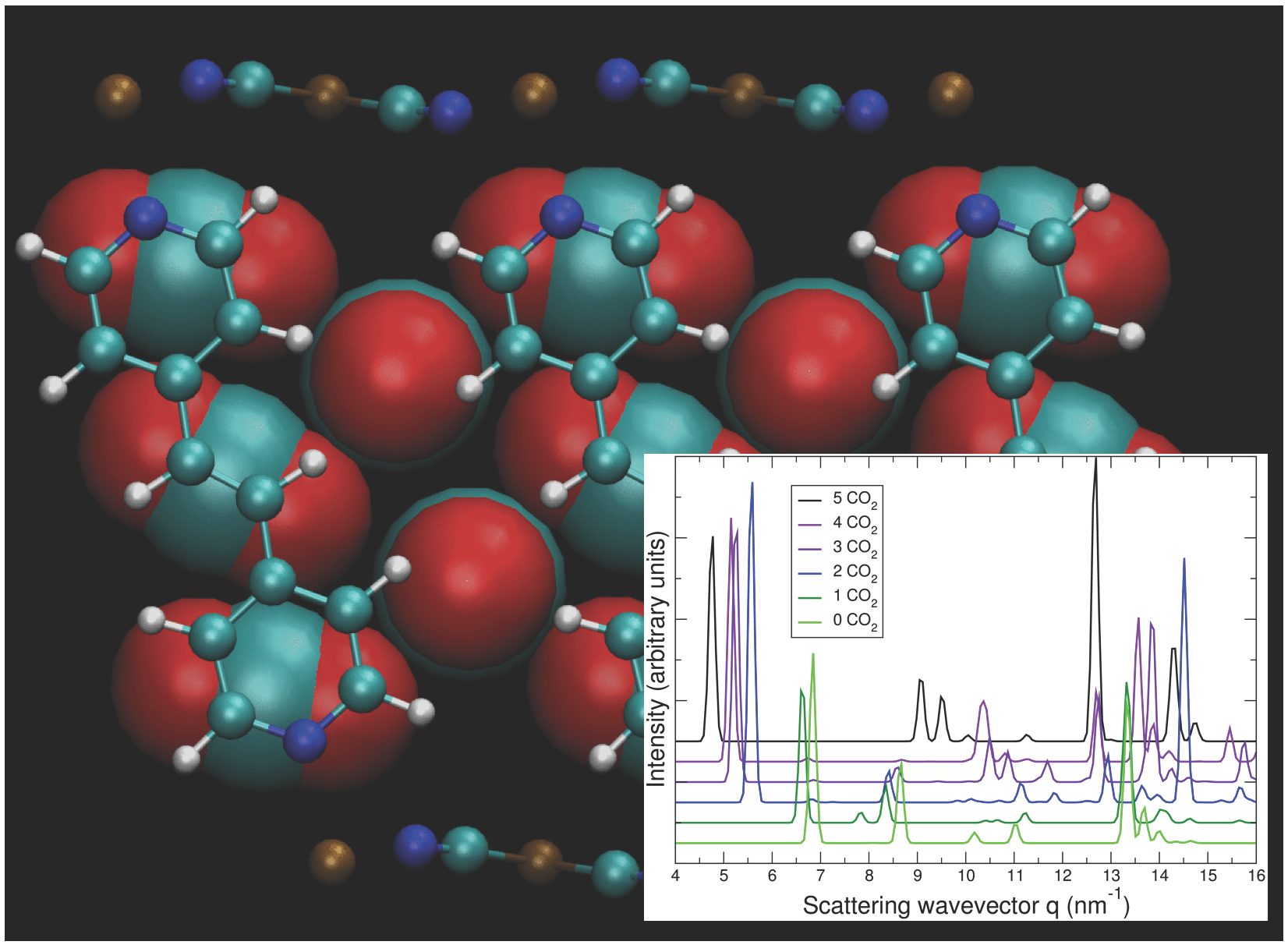
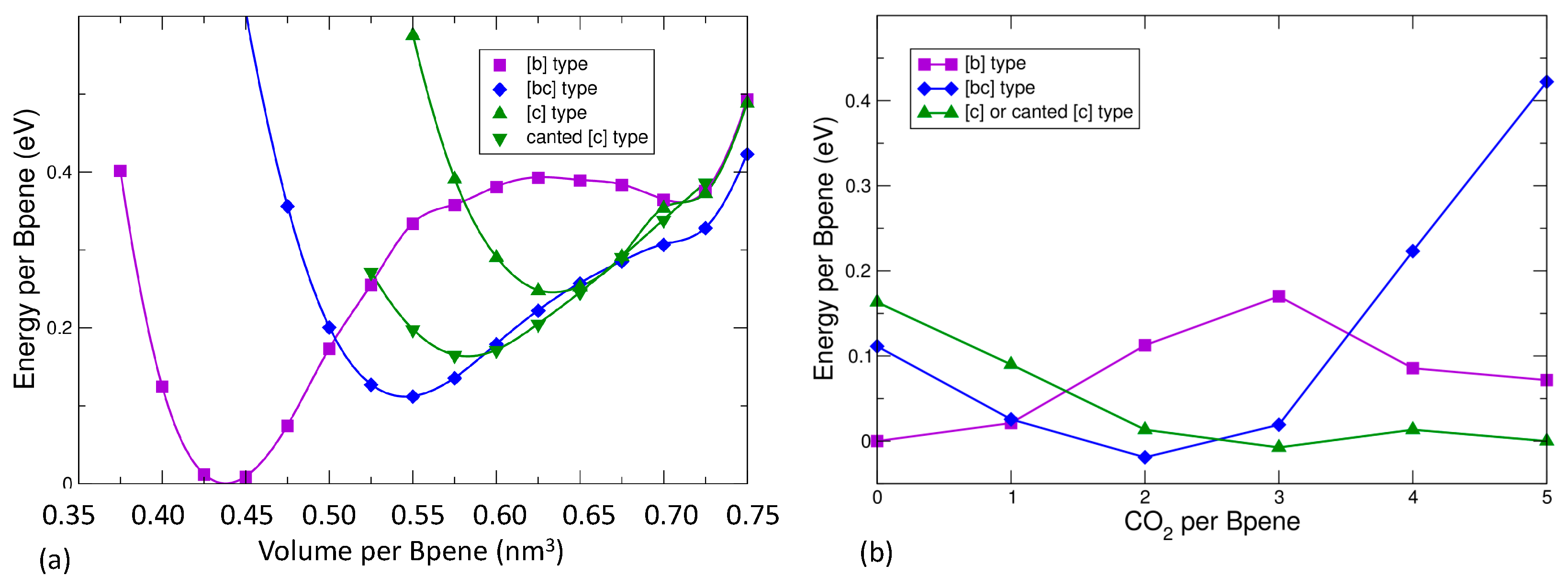
3.5. DFT Model Results for NiBpene
4. Concluding Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, M.W.; Chae, H.K.; Eddaourdi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-R.; Ma, Y.G.; McCarthy, M.C.; Sculley, J.; Yu, J.; Jeong, H.-K.; Balbuena, P.B. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011, 255, 1791–1823. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon dioxide capture in metal–organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, A.; Bon, V.; Schwedler, I.; Senkovska, I.; Kaskel, S.; Fischer, R.A. Flexible metal–organic frameworks. Chem. Soc. Rev. 2014, 43, 6062–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adil, K.; Belmabkhout, Y.; Pillai, R.S.; Cadiau, A.; Bhatt, P.M.; Assen, A.H.; Maurin, G.; Eddaoudi, M. Gas/vapor separation using ultra-microporous metal-organic frameworks: Insights into the structure/separation relationship. Chem. Soc. Rev. 2017, 46, 3402–3430. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Schubert, M.; Teich, F.; Puetter, H.; Schierle-Arndt, K.; Pastré, J. Metal-organic frameworks—Prospective industrial applications. J. Mater. Chem. 2006, 16, 626–636. [Google Scholar] [CrossRef]
- Zhu, Q.L.; Xu, Q. Metal-organic framework composites. Chem. Soc. Rev. 2014, 43, 5468–5512. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Mahmood, A.; Zou, R.Q.; Xu, Q. Metal-organic frameworks and their derived nanostructures for electrochemical energy storage and conversion. Energy Environ. Sci. 2015, 8, 1837–1866. [Google Scholar] [CrossRef]
- Mason, J.A.; Veenstra, M.; Long, J.R. Evaluating metal–organic frameworks for natural gas storage. Chem. Sci. 2014, 5, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Campbell, M.G.; Dinca, M. Electrically conductive porous metal-organic frameworks. Angew. Chem. Int. Ed. 2016, 55, 3566–3579. [Google Scholar] [CrossRef] [PubMed]
- Coudert, F.-X.; Jeffroy, M.; Alain, H.; Fuchs, M.A.; Boutin, A.; Mellot-Draznieks, C. Thermodynamics of guest-induced structural transitions in hybrid organic—Inorganic frameworks. J. Am. Chem. Soc. 2008, 130, 14294–14302. [Google Scholar] [CrossRef] [PubMed]
- Klein, N.; Hoffmann, H.C.; Cadiau, A.; Getzschmann, J.; Lohe, M.R.; Paasch, S.; Heydenreich, T.; Adil, K.; Senkovska, I.; Brunner, E.; et al. Structural flexibility and intrinsic dynamics in the M-2(2,6-ndc)(2)(dabco) (M = Ni, Cu, Co, Zn) metal-organic frameworks. J. Mater. Chem. 2012, 22, 10303–10312. [Google Scholar] [CrossRef]
- Mason, J.A.; Oktawiec, J.; Taylor, M.K.; Hudson, M.R.; Rodriguez, J.; Bachman, J.E.; Gonzalez, M.I.; Cervellino, A.; Guagliardi, A.; Brown, C.M.; et al. Methane storage in flexible metal–organic frameworks with intrinsic thermal management. Nature 2015, 527, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.H.; Liu, T.F.; Chen, Y.P.; Zou, L.F.; Feng, D.W.; Wang, K.C.; Zhang, Q.; Yuan, S.; Zhong, C.L.; Zhou, H.C. A Reversible crystallinity-preserving phase transition in metal-organic frameworks: Discovery, mechanistic studies, and potential applications. J. Am. Chem. Soc. 2015, 137, 7740–7746. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Wang, H.; Plonka, A.M.; Emge, T.J.; Parise, J.B.; Li, J. Direct structural identification of gas induced gate-opening coupled with commensurate adsorption in a microporous metal-organic framework. Chem. A Eur. J. 2016, 22, 11816–11825. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, D.; Nakagawa, K.; Higuchi, M.; Horike, S.; Kubota, Y.; Kobayashi, L.C.; Takata, M.; Kitagawa, S. Kinetic gate-opening process in a flexible porous coordination polymer. Angew. Chem. Int. Ed. 2008, 47, 3914–3918. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Matsuda, R.; Sakamoto, H.; Bonneau, C.; Kitagawa, S. A pillared-layer coordination polymer with a rotatable pillar acting as a molecular gate for guest molecules. J. Am. Chem. Soc. 2009, 131, 12792–12800. [Google Scholar] [CrossRef] [PubMed]
- Nijem, N.; Wu, H.H.; Canepa, P.; Marti, A.; Balkus, K.J.; Thonhauser, T.; Li, J.; Chabal, Y.J. Tuning the gate opening pressure of metal-organic frameworks (MOFs) for the selective separation of hydrocarbons. J. Am. Chem. Soc. 2012, 134, 15201–15204. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kotani, R.; Kondo, A.; Maeda, K. Structural investigation of a flexible MOF [Cu(BF4)(2)(1,3-bis(4-pyridyl)propane)(2)] showing selective gate adsorption with dynamic pore-opening/pore-closing processes. J. Phys. Chem. C 2016, 120, 21571–21579. [Google Scholar] [CrossRef]
- Elsaidi, S.K.; Mohamed, M.H.; Banerjee, D.; Thallapally, P.K. Flexibility in metal-organic frameworks: A fundamental understanding. Coord. Chem. Rev. 2018, 358, 125–152. [Google Scholar] [CrossRef]
- Yamada, K.; Tanaka, H.; Yagishita, S.; Adachi, K.; Uemura, T.; Kitagawa, S.; Kawata, S. Stepwise guest adsorption with large hysteresis in a coordination polymer {[Cu(bhnq)(THF)(2)](THF)}(n) constructed from a flexible hingelike ligand. Inorg. Chem. 2006, 45, 4322–4324. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Dinca, M.; Long, J.R. Broadly hysteretic H-2 adsorption in the microporous metal-organic framework Co(1,4-benzenedipyrazolate). J. Am. Chem. Soc. 2008, 130, 7848–7850. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.T.; Smith, M.R.; Bittner, E.; Bockrath, B. Hysteresis in the physisorption of CO2 and N2 in a flexible pillared layer nickel cyanide. J. Am. Chem. Soc. 2008, 130, 12427–12434. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Chun, H. Hysteretic gas sorption in a microporous metal-organic framework with nonintersecting 3D channels. Euro. J. Inorg. Chem. 2009, 33, 4946–4949. [Google Scholar] [CrossRef]
- Wu, H.H.; Thibault, C.G.; Wang, H.; Cychosz, K.A.; Thommes, M.; Li, J. Effect of temperature on hydrogen and carbon dioxide adsorption hysteresis in an ultramicroporous MOF. Microporous Mesoporous Mater. 2016, 219, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Ramsahye, N.A.; Trung, T.K.; Bourrelly, S.; Yang, Q.Y.; Devic, T.; Maurin, G.; Horcajada, P.; Llewellyn, P.L.; Yot, P.; Serre, C.; et al. Influence of the organic ligand functionalization on the breathing of the porous iron terephthalate metal-organic framework type material upon hydrocarbon adsorption. J. Phys. Chem. C 2011, 115, 18683–18695. [Google Scholar] [CrossRef]
- Mu, B.; Li, F.; Huang, Y.G.; Walton, K.S. Breathing effects of CO2 adsorption on a flexible 3D lanthanide metal-organic framework. J. Mater. Chem. 2012, 22, 10172–10178. [Google Scholar] [CrossRef]
- Chen, L.J.; Mowat, J.P.S.; Fairen-Jimenez, D.; Morrison, C.A.; Thompson, S.P.; Wright, P.A.; Duren, T. Elucidating the breathing of the metal-organic framework MIL-53(Sc) with ab Initio molecular dynamics simulations and in situ X-ray powder diffraction experiments. J. Am. Chem. Soc. 2013, 135, 15763–15773. [Google Scholar] [CrossRef] [PubMed]
- Lama, P.; Aggarwal, H.; Bezuidenhout, C.X.; Barbour, L.J. Giant hysteretic sorption of CO2: In situ crystallographic visualization of guest binding within a breathing framework at 298 K. Angew. Chem. Int. Ed. 2016, 55, 13271–13275. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.X.; Li, W.X.; Zhang, W.H.; Lang, J.P. Guest-induced switchable breathing behavior in a flexible metal-organic framework with pronounced negative gas pressure. Inorg. Chem. 2018, 57, 8627–8633. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Lin, X.; Lewis, W.; Suyetin, M.; Bichoutskaia, E.; Parker, J.E.; Tang, C.C.; Allan, D.R.; Rizkallah, P.J.; Hubberstey, P.; et al. A partially interpenetrated metal-organic framework for selective hysteretic sorption of carbon dioxide. Nat. Mater. 2012, 11, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.T.; Sui, L.; Goodman, A.; Luebke, D. Carbon dioxide (CO2) absorption behavior of mixed matrix polymer composites containing a flexible coordination polymer. J. Coll. Int. Sci. 2013, 393, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.M.; Braun, E.; Carraro, C.; Smit, B. Statistical mechanical model of gas adsorption in porous crystals with dynamic moieties. Proc. Natl. Acad. Sci. USA 2017, 114, E287–E296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.Y.; Li, Y.Z.; Jiang, C.Y.; Wang, H.H.; Hou, L.; Wang, Y.Y.; Zhu, Z.H. An interpenetrated pillar-layered metal-organic framework with novel clusters: Reversible structural transformation and selective gate-opening adsorption. Cryst. Growth Des. 2018, 18, 3044–3050. [Google Scholar] [CrossRef]
- Coudert, F.-X. Responsive metal–organic frameworks and framework materials: Under pressure, taking the heat, in the spotlight, with friends. Chem. Mater. 2015, 27, 1905–1916. [Google Scholar] [CrossRef]
- Bennett, T.D.; Cheetham, A.K. Amorphous metal-organic frameworks. Acc. Chem. Res. 2014, 47, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.L.; Culp, J.T.; Allen, A.J.; Espinal, L.; Wong-Ng, W.; Brown, T.D.; Goodman, A.; Bernardo, M.P.; Pancoast, R.J.; Chirdon, D.; et al. Selective adsorption of CO2 from light gas mixtures by using a structurally dynamic porous coordination polymer. Angew. Chem. Int. Ed. 2011, 50, 10888–10892. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.T.; Madden, C.; Kauffman, K.; Shi, F.; Matranga, C. Screening Hofmann compounds as CO2 sorbents: Nontraditional synthetic route to over 40 different pore-functionalized and flexible pillared cyanonickelates. Inorg. Chem. 2013, 52, 4205–4216. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ng, W.; Culp, J.T.; Chen, Y.S. Crystallography of representative MOFs based on pillared cyanonickelate (PICNIC) architecture. Crystals 2016, 6, 108. [Google Scholar] [CrossRef]
- Bury, W.; Fairen-Jimenez, D.; Lalonde, M.B.; Snurr, R.Q.; Farha, O.K.; Hupp, J.T. Control over catenation in pillared paddlewheel metal-organic framework materials via solvent-assisted linker exchange. Chem. Mater. 2013, 25, 739–744. [Google Scholar] [CrossRef]
- Luo, X.L.; Yin, Z.; Zeng, M.H.; Kurmoo, M. The construction, structures, and functions of pillared layer metal-organic frameworks. Inorg. Chem. Front. 2016, 3, 1208–1226. [Google Scholar] [CrossRef]
- Culp, J.T.; Natesakhawat, S.; Smith, M.R.; Bittner, E.; Matranga, C.; Bockrath, B. Hydrogen storage properties of rigid three-dimensional Hofmann clathrate derivatives: The effects of pore size. J. Phys. Chem. C 2008, 112, 7079–7083. [Google Scholar] [CrossRef]
- Wong-Ng, W.; Culp, J.T.; Chen, Y.S.; Zavalij, P.; Espinal, L.; Siderius, D.W.; Allen, A.J.; Scheins, S.; Matranga, C. Improved synthesis and crystal structure of the flexible pillared layer porous coordination polymer: Ni(1,2-bis(4-pyridyl)ethylene)[Ni(CN)(4)]. Crystengcom 2013, 15, 4684–4693. [Google Scholar] [CrossRef]
- Allen, A.J.; Espinal, L.; Wong-Ng, W.; Queen, W.L.; Brown, C.M.; Kline, S.R.; Kauffman, K.L.; Culp, J.T.; Matranga, C. Flexible metal-organic framework compounds: In situ studies for selective CO2 capture. J. Alloy. Compd. 2015, 647, 24–34. [Google Scholar] [CrossRef]
- Espinal, L.; Wong-Ng, W.; Kaduk, J.A.; Allen, A.J.; Snyder, C.R.; Chiu, C.; Siderius, D.W.; Li, L.; Cockayne, E.; Espinal, A.E.; et al. Time-dependent CO2 sorption hysteresis in a one-dimensional microporous octahedral molecular sieve. J. Am. Chem. Soc. 2012, 134, 7944–7951. [Google Scholar] [CrossRef] [PubMed]
- Ilavsky, J.; Jemian, P.R.; Allen, A.J.; Zhang, F.; Levine, L.E.; Long, G.G. Ultra-small-angle X-ray scattering at the Advanced Photon Source. J. Appl. Cryst. 2009, 42, 469–479. [Google Scholar] [CrossRef]
- Ilavsky, J.; Zhang, F.; Allen, A.J.; Levine, L.E.; Jemian, P.R.; Long, G.G. Ultra-small-angle X-ray scattering instrument at the Advanced Photon Source: History, recent development, and current status. Metall. Mater. Trans. A 2013, 44, 68–76. [Google Scholar] [CrossRef]
- Ilavsky, J.; Zhang, F.; Andrews, R.N.; Kuzmenko, I.; Jemian, P.R.; Levine, L.E.; Allen, A.J. Development of combined microstructure and structure characterization facility for in situ and operando studies at the Advanced Photon Source. J. Appl. Cryst. 2018, 51, 867–882. [Google Scholar] [CrossRef]
- Hendon, C.H.; Rieth, A.J.; Korzynski, M.D.; Dinca, M. Grand challenges and future opportunities for metal-organic frameworks. ACS Cent. Sci. 2017, 3, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal-organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Glinka, C.J.; Barker, J.G.; Hammouda, B.; Krueger, S.; Moyer, J.J.; Orts, W.J. The 30m SANS instruments at NIST. J. Appl. Cryst. 1998, 31, 430–445. [Google Scholar] [CrossRef]
- Kline, S.R. Reduction and analysis of SANS and USANS data using IGOR Pro. J. Appl. Cryst. 2006, 39, 895–900. [Google Scholar] [CrossRef]
- Black, D.R.; Windover, D.; Henins, A.; Filliben, J.; Cline, J.P. Certification of Standard Reference Material 660B. Powder Diffr. 2011, 26, 155–158. [Google Scholar] [CrossRef]
- Ilavsky, J.; Jemian, P.R. Irena: Tool suite for modeling and analysis of small-angle scattering. J. Appl. Cryst. 2009, 42, 347–353. [Google Scholar] [CrossRef]
- Ilavsky, J. Nika: Software for two-dimensional data reduction. J. Appl. Cryst. 2012, 45, 324–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K.; Tao, J.; Staroverov, V.N.; et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [Green Version]
- Cockayne, E. Contribution of density functional theory to microporous materials for carbon capture. In Materials and Processes for CO2 Capture, Conversion, and Sequestration; Li, L., Wong-Ng, W., Huang, K., Cook, L.P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 319–343. ISBN 9781119231035. [Google Scholar] [CrossRef]
- Ortiz, A.U.; Boutin, A.; Fuchs, A.H.; Coudert, F.-X. Anisotropic Elastic Properties of Flexible Metal-Organic Frameworks: How Soft are Soft Porous Crystals? Phys. Rev. Lett. 2012, 109, 195502. [Google Scholar] [CrossRef] [PubMed]
- Cockayne, E. Thermodynamics of the Flexible Metal–Organic Framework Material MIL-53(Cr) From First-Principles. J. Phys. Chem. C 2017, 121, 4312–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Improved density dependent correction for the description of London dispersion forces. J. Chem. Theory Comput. 2013, 9, 4293–4299. [Google Scholar] [CrossRef] [PubMed]
- Bucko, T.; Lebègue, S.; Ángyán, J.G.; Hafner, J. Extending the applicability of the Tkatchenko-Scheffler dispersion correction via iterative Hirshfeld partitioning. J. Chem. Phys. 2014, 141, 034114. [Google Scholar] [CrossRef] [PubMed]
- Bultinck, P.; Van Alsenoy, C.; Ayers, P.W.; Carbó Dorca, R. Critical analysis and extension of the Hirshfeld atoms in molecules. J. Chem. Phys. 2007, 126, 144111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed]
- Span, R.; Wagner, W. A new equation of state for carbon dioxide covering the fluid region from the triple-point temperature to 1100K at pressures up to 800 MPa. J. Phys. Chem. Ref. Data 1996, 25, 1509–1596. [Google Scholar] [CrossRef]
- Siderius, D.W.; Gelb, L.D. Predicting Gas Adsorption in Complex Microporous and Mesoporous Materials Using a New Density Functional Theory of Finely Discretized Lattice Fluids. Langmuir 2009, 25, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Ghysels, A.; Vanduyfhuys, L.; Vandichel, M.; Waroquier, M.; Van Speybroeck, V.; Smit, B. On the Thermodynamics of Framework Breathing: A Free Energy Model for Gas Adsorption in MIL-53. J. Phys. Chem. C 2013, 117, 11450–11554. [Google Scholar] [CrossRef]
- Freund, R.; Lachelt, U.; Gruber, T.; Ruhle, B.; Wuttke, S. Multifunctional Efficiency: Extending the Concept of Atom Economy to Functional Nanomaterials. ACS Nano 2018, 12, 2094–2105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, Y.; Yang, J.; Diercks, C.S.; Tamura, N.; Jin, F.; Yaghi, O.M. Molecular Weaving of Covalent Organic Frameworks for Adaptive Guest Inclusion. J. Am. Chem. Soc. 2018, 140, 16015–16019. [Google Scholar] [CrossRef] [PubMed]
- Modena, M.M.; Hirschle, P.; Wuttke, S.; Burg, T.P. Porous Nanoparticles: Mass Measurements Reveal Preferential Sorption of Mixed Solvent Components in Porous Nanoparticles. Small 2018, 14, 1800826. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.M.; Mason, J.A.; Kong, X.; Bloch, E.D.; Gygi, D.; Dani, A.; Crocellà, V.; Giordanino, F.; Odoh, S.O.; Drisdell, W.S.; et al. Cooperative Insertion of CO2 in Diamine-Appended Metal-Organic Frameworks. Nature 2015, 519, 303–308. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | T (°C) | (bar) | a (nm) | b (nm) | c (nm) | β (°) | V (nm3) |
|---|---|---|---|---|---|---|---|
| 1 | 90 | 80 | 1.373(2) 1 | 1.447(4) | 1.3513(9) | 107.06(9) | 2.556(8) |
| 2 | 90 | 73 | 1.3647(9) | 1.4353(7) | 1.3522(15) | 107.45(10) | 2.527(4) |
| 3 | 90 | 70 | 1.3683(15) | 1.4301(6) | 1.3469(9) | 108.43(7) | 2.500(3) |
| 4 | 90 | 2 | 1.3597(6) | 1.442(2) | 1.356(3) | 114.22(6) | 2.425(6) |
| 5 | 60 | 10 | 1.357(4) | 1.4087(12) | 1.332(3) | 114.6(2) | 2.314(9) |
| 6 | 60 | 2 | 1.360(3) | 1.4080(9) | 1.327(2) | 115.92(15) | 2.285(6) |
| 7 | 30 | 80 | 1.3616(9) | 1.452(4) | 1.344(2) | 107.92(9) | 2.527(8) |
| 8 | 30 | 65 | 1.357(3) | 1.437(2) | 1.338(3) | 108.7(3) | 2.471(8) |
| 9 | 25 | 0 | 1.3529(1) | 1.3966(2) | 1.321(2) | 116.66(6) | 2.231(4) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, A.J.; Wong-Ng, W.; Cockayne, E.; Culp, J.T.; Matranga, C. Structural Basis of CO2 Adsorption in a Flexible Metal-Organic Framework Material. Nanomaterials 2019, 9, 354. https://doi.org/10.3390/nano9030354
Allen AJ, Wong-Ng W, Cockayne E, Culp JT, Matranga C. Structural Basis of CO2 Adsorption in a Flexible Metal-Organic Framework Material. Nanomaterials. 2019; 9(3):354. https://doi.org/10.3390/nano9030354
Chicago/Turabian StyleAllen, Andrew J., Winnie Wong-Ng, Eric Cockayne, Jeffrey T. Culp, and Christopher Matranga. 2019. "Structural Basis of CO2 Adsorption in a Flexible Metal-Organic Framework Material" Nanomaterials 9, no. 3: 354. https://doi.org/10.3390/nano9030354