CO2 Hydrogenation over Nanoceria-Supported Transition Metal Catalysts: Role of Ceria Morphology (Nanorods versus Nanocubes) and Active Phase Nature (Co versus Cu)

 ,
, 

 ,
,  ,
,
Abstract
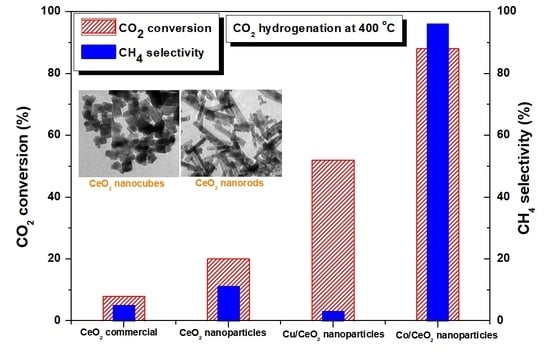
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Materials Synthesis
2.3. Materials Characterization
2.4. Catalytic Evaluation Studies
3. Results and Discussion
3.1. Morphological Characterization (TEM)
3.2. Textural and Structural Characterization (Brunauer–Emmett–Teller (BET), X-ray Diffraction (XRD))
3.3. Redox Properties (Hydrogen Temperature-Programmed Reduction (H2-TPR))
3.4. Catalytic Evaluation Studies
3.4.1. CO2 Hydrogenation Activity
3.4.2. Effect of H2:CO2 Ratio
3.4.3. Stability Tests
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic chemical carbon cycle for a sustainable future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.A.; Liu, P.; Stacchiola, D.J.; Senanayake, S.D.; White, M.G.; Chen, J.G. Hydrogenation of CO2 to Methanol: Importance of Metal-Oxide and Metal-Carbide Interfaces in the Activation of CO2. ACS Catal. 2015, 5, 6696–6706. [Google Scholar] [CrossRef]
- Xu, X.; Moulijn, J.A. Mitigation of CO2 by chemical conversion: Plausible chemical reactions and promising products. Energy Fuels 1996, 10, 305–325. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Sabatier, P.; Senderens, J.B. New Synthesis of Methane. Comptes Rendus Hebd. Seances Acad. Scrences 1902, 134, 514–516. [Google Scholar]
- Bahruji, H.; Bowker, M.; Hutchings, G.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.J.; Zeng, Z.; Liao, F.; Hong, X.; Tsang, S.C.E. Enhanced CO2 hydrogenation to methanol over CuZn nanoalloy in Ga modified Cu/ZnO catalysts. J. Catal. 2016, 343, 157–167. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Ramírez, P.J.; Waluyo, I.; Kundu, S.; Mudiyanselage, K.; Liu, Z.; Liu, Z.; Axnanda, S.; Stacchiola, D.J.; Evans, J.; et al. Hydrogenation of CO2 to Methanol on CeOx/Cu(111) and ZnO/Cu(111) Catalysts: Role of the Metal-Oxide Interface and Importance of Ce3+ Sites. J. Phys. Chem. C 2016, 120, 1778–1784. [Google Scholar] [CrossRef] [Green Version]
- Vourros, A.; Garagounis, I.; Kyriakou, V.; Carabineiro, S.A.C.; Maldonado-Hódar, F.J.; Marnellos, G.E.; Konsolakis, M. Carbon dioxide hydrogenation over supported Au nanoparticles: Effect of the support. J. CO2 Util. 2017, 19, 247–256. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Ereña, J.; Sierra, I.; Olazar, M.; Bilbao, J. Deactivation and regeneration of hybrid catalysts in the single-step synthesis of dimethyl ether from syngas and CO2. Catal. Today 2005, 106, 265–270. [Google Scholar] [CrossRef]
- Zhang, Y.; Fei, J.; Yu, Y.; Zheng, X. Silica immobilized ruthenium catalyst used for carbon dioxide hydrogenation to formic acid (I): The effect of functionalizing group and additive on the catalyst performance. Catal. Commun. 2004, 5, 643–646. [Google Scholar] [CrossRef]
- Saeidi, S.; Amin, N.A.S.; Rahimpour, M.R. Hydrogenation of CO2 to value-added products—A review and potential future developments. J. CO2 Util. 2014, 5, 66–81. [Google Scholar] [CrossRef]
- Kaiser, P.; Unde, R.B.; Kern, C.; Jess, A. Production of liquid hydrocarbons with CO2 as carbon source based on reverse water-gas shift and Fischer-Tropsch synthesis. Chem. Ing. Tech. 2013, 85, 489–499. [Google Scholar] [CrossRef]
- Pastor-Pérez, L.; Baibars, F.; Le Sache, E.; Arellano-García, H.; Gu, S.; Reina, T.R. CO2 valorisation via Reverse Water-Gas Shift reaction using advanced Cs doped Fe-Cu/Al2O3 catalysts. J. CO2 Util. 2017, 21, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Rafiee, A.; Rajab Khalilpour, K.; Milani, D.; Panahi, M. Trends in CO2 conversion and utilization: A review from process systems perspective. J. Environ. Chem. Eng. 2018, 6, 5771–5794. [Google Scholar] [CrossRef]
- Tada, S.; Ochieng, O.J.; Kikuchi, R.; Haneda, T.; Kameyama, H. Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts. Int. J. Hydrog. Energy 2014, 39, 10090–10100. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Mazza, A.; Bompard, E.; Chicco, G. Applications of power to gas technologies in emerging electrical systems. Renew. Sustain. Energy Rev. 2018, 92, 794–806. [Google Scholar] [CrossRef]
- Lewandowska-Bernat, A.; Desideri, U. Opportunities of Power-to-Gas technology. Energy Procedia 2017, 105, 4569–4574. [Google Scholar] [CrossRef]
- Van Leeuwen, C.; Mulder, M. Power-to-gas in electricity markets dominated by renewables. Appl. Energy 2018, 232, 258–272. [Google Scholar] [CrossRef]
- Götz, M.; Lefebvre, J.; Mörs, F.; McDaniel Koch, A.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A technological and economic review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef] [Green Version]
- Ghaib, K.; Ben-Fares, F.Z. Power-to-Methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018, 81, 433–446. [Google Scholar] [CrossRef]
- Zangeneh, F.T.; Sahebdelfar, S.; Ravanchi, M.T. Conversion of carbon dioxide to valuable petrochemicals: An approach to clean development mechanism. J. Nat. Gas Chem. 2011, 20, 219–231. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Fazlollahi, F.; Nikoo, M.K.; Sefidkon, F.; Klemeš, J.J.; Baxter, L.L. Mechanisms and kinetics of CO2 hydrogenation to value-added products: A detailed review on current status and future trends. Renew. Sustain. Energy Rev. 2017, 80, 1292–1311. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Heterogeneous catalytic reactions with CO2: Status and perspectives. Stud. Surf. Sci. Catal. 2004, 153, 1–8. [Google Scholar] [CrossRef]
- Boaro, M.; Colussi, S.; Trovarelli, A. Ceria-based materials in hydrogenation and reforming reactions for CO2 valorization. Front. Chem. 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Ghaib, K.; Nitz, K.; Ben-Fares, F.Z. Chemical Methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Scibioh, M.A.; Viswanathan, B. Chapter 5—Heterogeneous Hydrogenation of CO2. Carbon Dioxide Chem. Fuels 2018, 191–253. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Winter, L.R.; Chen, R.; Chen, X.; Chang, K.; Liu, Z.; Senanayake, S.D.; Ebrahim, A.M.; Chen, J.G. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B Environ. 2019, 245, 360–366. [Google Scholar] [CrossRef]
- Melchionna, M.; Fornasiero, P. The role of ceria-based nanostructured materials in energy applications. Mater. Today 2014, 17, 349–357. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha, P.; Bhagiyalakshmi, M.; Ganesh, M.; Palanichamy, M.; Murugesan, V.; Jang, H.T. Role of ceria in CO2 adsorption on NaZSM-5 synthesized using rice husk ash. J. Ind. Eng. Chem. 2012, 18, 260–265. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Fornasiero, P.; Gorte, R.J. Opportunities for tailoring catalytic properties through metal-support interactions. Catal. Lett. 2012, 142, 1043–1048. [Google Scholar] [CrossRef]
- Konsolakis, M. The role of Copper–Ceria interactions in catalysis science: Recent theoretical and experimental advances. Appl. Catal. B Environ. 2016, 198, 49–66. [Google Scholar] [CrossRef]
- Sharma, S.; Hu, Z.; Zhang, P.; McFarland, E.W.; Metiu, H. CO2 methanation on Ru-doped ceria. J. Catal. 2011, 278, 297–309. [Google Scholar] [CrossRef]
- Beuls, A.; Swalus, C.; Jacquemin, M.; Heyen, G.; Karelovic, A.; Ruiz, P. Methanation of CO2: Further insight into the mechanism over Rh/γ-Al2O3 catalyst. Appl. Catal. B Environ. 2012, 113–114, 2–10. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Valverde, J.L.; Sánchez, P.; Dorado, F. CO2 Hydrogenation to Methanol at Atmospheric Pressure: Influence of the Preparation Method of Pd/ZnO Catalysts. Catal. Lett. 2016, 146, 373–382. [Google Scholar] [CrossRef]
- Spezzati, G.; Benavidez, A.D.; DeLaRiva, A.T.; Su, Y.; Hofmann, J.P.; Asahina, S.; Olivier, E.J.; Neethling, J.H.; Miller, J.T.; Datye, A.K.; et al. CO oxidation by Pd supported on CeO2(100) and CeO2(111) facets. Appl. Catal. B Environ. 2019, 243, 36–46. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakeimidis, Z. Surface/structure functionalization of copper-based catalysts by metal-support and/or metal-metal interactions. Appl. Surf. Sci. 2014, 320, 244–255. [Google Scholar] [CrossRef]
- Park, J.B.; Graciani, J.; Evans, J.; Stacchiola, D.; Senanayake, S.D.; Barrio, L.; Liu, P.; Sanz, J.F.; Hrbek, J.; Rodriguez, J.A. Gold, copper, and platinum nanoparticles dispersed on CeOx/TiO2(110) surfaces: High water-gas shift activity and the nature of the mixed-metal oxide at the nanometer level. J. Am. Chem. Soc. 2010, 132, 356–363. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.E.; Plucinski, P.; Mattia, D.; Torrente-Murciano, L.; Ting, V.P.; Jones, M.D. Effect of support of Co-Na-Mo catalysts on the direct conversion of CO2 to hydrocarbons. J. CO2 Util. 2016, 16, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Le, T.A.; Kim, T.W.; Lee, S.H.; Park, E.D. Effects of Na content in Na/Ni/SiO2 and Na/Ni/CeO2 catalysts for CO and CO2 methanation. Catal. Today 2018, 303, 159–167. [Google Scholar] [CrossRef]
- Sun, C.; Li, H.; Chen, L. Nanostructured ceria-based materials: Synthesis, properties, and applications. Energy Environ. Sci. 2012, 5, 8475–8505. [Google Scholar] [CrossRef]
- Yao, X.; Tang, C.; Gao, F.; Dong, L. Research progress on the catalytic elimination of atmospheric molecular contaminants over supported metal-oxide catalysts. Catal. Sci. Technol. 2014, 4, 2814–2829. [Google Scholar] [CrossRef]
- Ganduglia-Pirovano, M.V.; Hofmann, A.; Sauer, J. Oxygen vacancies in transition metal and rare earth oxides: Current state of understanding and remaining challenges. Surf. Sci. Rep. 2007, 62, 219–270. [Google Scholar] [CrossRef]
- Ouyang, B.; Tan, W.; Liu, B. Morphology effect of nanostructure ceria on the Cu/CeO2 catalysts for synthesis of methanol from CO2 hydrogenation. Catal. Commun. 2017, 95, 36–39. [Google Scholar] [CrossRef]
- Si, R.; Flytzani-Stephanopoulos, M. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 catalysts for the water-gas shift reaction. Angew. Chem. Int. Ed. 2008, 47, 2884–2887. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Xu, H.; Han, Y. Reverse water-gas shift reaction over ceria nanocube synthesized by hydrothermal method. Catal. Commun. 2016, 76, 1–6. [Google Scholar] [CrossRef]
- Yang, S.C.; Pang, S.H.; Sulmonetti, T.P.; Su, W.N.; Lee, J.F.; Hwang, B.J.; Jones, C.W. Synergy between Ceria Oxygen Vacancies and Cu Nanoparticles Facilitates the Catalytic Conversion of CO2 to CO under Mild Conditions. ACS Catal. 2018, 8, 12056–12066. [Google Scholar] [CrossRef]
- Muroyama, H.; Tsuda, Y.; Asakoshi, T.; Masitah, H.; Okanishi, T.; Matsui, T.; Eguchi, K. Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J. Catal. 2016, 343, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Le, T.A.; Kim, M.S.; Lee, S.H.; Kim, T.W.; Park, E.D. CO and CO2 methanation over supported Ni catalysts. Catal. Today 2017, 293–294, 89–96. [Google Scholar] [CrossRef]
- Aldana, P.A.U.; Ocampo, F.; Kobl, K.; Louis, B.; Thibault-Starzyk, F.; Daturi, M.; Bazin, P.; Thomas, S.; Roger, A.C. Catalytic CO2 valorization into CH4 on Ni-based ceria-zirconia. Reaction mechanism by operando IR spectroscopy. Catal. Today 2013, 215, 201–207. [Google Scholar] [CrossRef]
- Martin, N.M.; Velin, P.; Skoglundh, M.; Bauer, M.; Carlsson, P.A. Catalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts. Catal. Sci. Technol. 2017, 7, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.M.; Hemmingsson, F.; Schaefer, A.; Ek, M.; Merte, L.R.; Hejral, U.; Gustafson, J.; Skoglundh, M.; Dippel, A.C.; Gutowski, O.; et al. Structure-function relationship for CO2 methanation over ceria supported Rh and Ni catalysts under atmospheric pressure conditions. Catal. Sci. Technol. 2019, 9, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Wei, S.; Zhou, S.; Zhang, T.; Wang, Z.; Ng, S.P.; Lu, X.; Wu, C.M.L.; Guo, W. Initial Reduction of CO2 on Pd-, Ru-, and Cu-Doped CeO2(111) Surfaces: Effects of Surface Modification on Catalytic Activity and Selectivity. ACS Appl. Mater. Interfaces 2017, 9, 26107–26117. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Carabineiro, S.A.C.; Iliopoulou, E.; Andriopoulou, C.; Kallithrakas-Kontos, N.; Boghosian, S.; Konsolakis, M. Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts. Appl. Catal. B Environ. 2018, 230, 18–28. [Google Scholar] [CrossRef]
- Sharma, V.; Eberhardt, K.M.; Sharma, R.; Adams, J.B.; Crozier, P.A. A spray drying system for synthesis of rare-earth doped cerium oxide nanoparticles. Chem. Phys. Lett. 2010, 495, 280–286. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, M.; Zhou, R. Effect of interaction between CuO and CeO2 on the performance of CuO-CeO2 catalysts for selective oxidation of CO in H2-rich streams. Indian J. Chem. 2012, 51, 1529–1537. [Google Scholar]
- Luo, J.Y.; Meng, M.; Li, X.; Li, X.G.; Zha, Y.Q.; Hu, T.D.; Xie, Y.N.; Zhang, J. Mesoporous Co3O4-CeO2 and Pd/Co3O4-CeO2 catalysts: Synthesis, characterization and mechanistic study of their catalytic properties for low-temperature CO oxidation. J. Catal. 2008, 254, 310–324. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Djinović, P.; Tchernychova, E.; Tkachenko, O.P.; Kustov, L.M.; Pintar, A. Nanoshaped CuO/CeO2 Materials: Effect of the Exposed Ceria Surfaces on Catalytic Activity in N2O Decomposition Reaction. ACS Catal. 2015, 5, 5357–5365. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakimidis, Z.; Kraia, T.; Marnellos, G.E. Hydrogen Production by Ethanol Steam Reforming (ESR) over CeO2 Supported Transition Metal (Fe, Co, Ni, Cu) Catalysts: Insight into the Structure-Activity Relationship. Catalysts 2016, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Gamarra, D.; Lόpez Cámara, A.; Monte, M.; Rasmussen, S.B.; Chinchilla, L.E.; Hungría, A.B.; Munuera, G.; Gyorffy, N.; Schay, Z.; Cortés Corberán, V.; et al. Preferential oxidation of CO in excess H2 over CuO/CeO2 catalysts: Characterization and performance as a function of the exposed face present in the CeO2 support. Appl. Catal. B Environ. 2013, 130–131, 224–238. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Sadowski, J.T.; Evans, J.; Kundu, S.; Agnoli, S.; Yang, F.; Stacchiola, D.; Flege, J.I.; Hrbek, J.; Rodriguez, J.A. Nanopattering in CeOx/Cu(111): A New Type of Surface Reconstruction and Enhancement of Catalytic Activity. J. Phys. Chem. Lett. 2012, 3, 839–843. [Google Scholar] [CrossRef]
- Lykaki, M.; Pachatouridou, E.; Iliopoulou, E.; Carabineiro, S.A.C.; Konsolakis, M. Impact of the synthesis parameters on the solid state properties and the CO oxidation performance of ceria nanoparticles. RSC Adv. 2017, 7, 6160–6169. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Huang, H.H.; Tang, C.W.; Wang, C.B. The effect of accessible oxygen over Co3O4-CeO2 catalysts on the steam reforming of ethanol. Int. J. Hydrog. Energy 2014, 39, 20700–20711. [Google Scholar] [CrossRef]
- Bayram, B.; Soykal, I.I.; Von Deak, D.; Miller, J.T.; Ozkan, U.S. Ethanol steam reforming over Co-based catalysts: Investigation of cobalt coordination environment under reaction conditions. J. Catal. 2011, 284, 77–89. [Google Scholar] [CrossRef]
- Aboukaïs, A.; Skaf, M.; Hany, S.; Cousin, R.; Aouad, S.; Labaki, M.; Abi-Aad, E. A comparative study of Cu, Ag and Au doped CeO2 in the total oxidation of volatile organic compounds (VOCs). Mater. Chem. Phys. 2016, 177, 570–576. [Google Scholar] [CrossRef]
- Jampa, S.; Jamieson, A.M.; Chaisuwan, T.; Luengnaruemitchai, A.; Wongkasemjit, S. Achievement of hydrogen production from autothermal steam reforming of methanol over Cu-loaded mesoporous CeO2 and Cu-loaded mesoporous CeO2–ZrO2 catalysts. Int. J. Hydrog. Energy 2017, 42, 15073–15084. [Google Scholar] [CrossRef]
- Kovacevic, M.; Mojet, B.L.; Van Ommen, J.G.; Lefferts, L. Effects of Morphology of Cerium Oxide Catalysts for Reverse Water Gas Shift Reaction. Catal. Lett. 2016, 146, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; Zhou, G.; Ge, S.; Xie, H.; Jiao, Z.; Zhang, G.; Xiong, K. CO2 reverse water-gas shift reaction on mesoporous M-CeO2 catalysts. Can. J. Chem. Eng. 2017, 95, 634–642. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castañeda, R.; Lin, J.; Senanayake, S.D.; et al. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D. Study of bimetallic Cu-Ni/γ-Al2O3 catalysts for carbon dioxide hydrogenation. Int. J. Hydrog. Energy 1999, 24, 351–354. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Study of iron-promoted Cu/SiO2 catalyst on high temperature reverse water gas shift reaction. Appl. Catal. A Gen. 2004, 257, 97–106. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.; Lin, L.; Yao, S.; Zhang, M.; Liu, X.; Wang, X.; Li, Y.W.; Shi, C.; Ma, D. Highly Dispersed Copper over β-Mo2C as an Efficient and Stable Catalyst for the Reverse Water Gas Shift (RWGS) Reaction. ACS Catal. 2017, 7, 912–918. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, H.; Xing, Y.; Xu, S.; Xie, H.; Xiong, K. CO2 hydrogenation to methane over mesoporous Co/SiO2 catalysts: Effect of structure. J. CO2 Util. 2018, 26, 221–229. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, T.; Xie, H.; Zheng, X. Effects of structure on the carbon dioxide methanation performance of Co-based catalysts. Int. J. Hydrog. Energy 2013, 38, 10012–10018. [Google Scholar] [CrossRef]
- Zhu, H.; Razzaq, R.; Li, C.; Muhmmad, Y.; Zhang, S. Catalytic Methanation of Carbon Dioxide by Active Oxygen Material CexZr1-xO2 Supported Ni-Co Bimetallic Nanocatalysts. AIChE J. 2013, 59, 2567–2576. [Google Scholar] [CrossRef]
- Suslova, E.V.; Chernyak, S.A.; Egorov, A.V.; Savilov, S.V.; Lunin, V.V. CO2 Hydrogenation over Cobalt-Containing Catalysts. Kinet. Catal. 2015, 56, 646–654. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Sánchez, P.; Kyriakou, V.; Zafeiratos, S.; Marnellos, G.E.; Konsolakis, M.; Dorado, F. Effect of support nature on the cobalt-catalyzed CO2 hydrogenation. J. CO2 Util. 2017, 21, 562–571. [Google Scholar] [CrossRef]
- Jwa, E.; Lee, S.B.; Lee, H.W.; Mok, Y.S. Plasma-assisted catalytic methanation of CO and CO2 over Ni-zeolite catalysts. Fuel Process. Technol. 2013, 108, 89–93. [Google Scholar] [CrossRef]
- Miao, B.; Ma, S.S.K.; Wang, X.; Su, H.; Chan, S.H. Catalysis mechanisms of CO2 and CO methanation. Catal. Sci. Technol. 2016, 6, 4048–4058. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni@MOF-5 via control of active species dispersion. Chem. Commun. 2015, 51, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, G.D.; Bartholomew, C.H. Hydrogenation of CO2 on Group VIII metals: II. Kinetics and Mechanism of CO2 Hydrogenation on Nickel. J. Catal. 1982, 77, 460–472. [Google Scholar] [CrossRef]
- Ginés, M.J.L.; Marchi, A.J.; Apesteguía, C.R. Kinetic study of the reverse water-gas shift reaction over CuO/ZnO/Al2O3 catalysts. Appl. Catal. A Gen. 1997, 154, 155–171. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H. Study on the Mechanism of CO Formation in Reverse Water Gas Shift Reaction Over Cu/SiO2 Catalyst by Pulse Reaction, TPD and TPR. Catal. Lett. 2002, 83, 121–126. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Zhang, X.; Wei, M.; Evans, D.G.; Duan, X. Catalytic behavior of supported Ru nanoparticles on the {100}, {110}, and {111} facet of CeO2. J. Catal. 2015, 329, 177–186. [Google Scholar] [CrossRef]
- Sahebdelfar, S.; Takht Ravanchi, M. Carbon dioxide utilization for methane production: A thermodynamic analysis. J. Pet. Sci. Eng. 2015, 134, 14–22. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Reiter, G.; Lindorfer, J. Global warming potential of hydrogen and methane production from renewable electricity via power-to-gas technology. Int. J. Life Cycle Assess. 2015, 20, 477–489. [Google Scholar] [CrossRef]
- Swapnesh, A.; Srivastava, V.C.; Mall, I.D. Comparative study on thermodynamic analysis of CO2 utilization reactions. Chem. Eng. Technol. 2014, 37, 1765–1777. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Study of reverse water gas shift reaction by TPD, TPR and CO2 hydrogenation over potassium-promoted Cu/SiO2 catalyst. Appl. Catal. A Gen. 2003, 238, 55–67. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yang, X.; Boscoboinik, J.A.; Chen, J.G. Molybdenum carbide as alternative catalysts to precious metals for highly selective reduction of CO2 to CO. Angew. Chem. Int. Ed. 2014, 53, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pastor-Pérez, L.; Gu, S.; Sepúlveda-Escribano, A.; Reina, T.R. Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: Effect of selected transition metal promoters. Appl. Catal. B Environ. 2018, 232, 464–471. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Enhanced activity and stability of a Cu/SiO2 catalyst for the reverse water gas shift reaction by an iron promoter. Chem. Commun. 2001, 1770–1771. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kubota, T.; Gotoh, H.; Ohto, Y.; Aritani, H.; Tanaka, T.; Yoshida, S. XAFS study of zirconia-supported copper catalysts for the NO-CO reaction: Deactivation, rejuvenation and stabilization of Cu species. J. Chem. Soc. Faraday Trans. 1998, 94, 3743–3752. [Google Scholar] [CrossRef]
- Li, J.; Zhou, L.; Zhu, Q.; Li, H. Enhanced methanation over aerogel NiCo/Al2O3 catalyst in a magnetic fluidized bed. Ind. Eng. Chem. Res. 2013, 52, 6647–6654. [Google Scholar] [CrossRef]
- Li, J.; Zhou, L.; Li, P.; Zhu, Q.; Gao, J.; Gu, F.; Su, F. Enhanced fluidized bed methanation over a Ni/Al2O3 catalyst for production of synthetic natural gas. Chem. Eng. J. 2013, 219, 183–189. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | BET Analysis | XRD Analysis | H2-TPR Analysis | ||||
|---|---|---|---|---|---|---|---|
| SBET (m2/g) | Average Crystallite Diameter, DXRD (nm) | H2 Consumption (mmol H2 g−1) 1 | Theoretical H2 (mmol H2 g−1) 2 | Peak Temperature (°C) | |||
| CeO2 | Co3O4/CuO | ||||||
| CeO2-NC | 37 | 27 | - | 0.41 | - | 589 | 809 |
| CeO2-NR | 79 | 15 | - | 0.59 | - | 545 | 788 |
| Co/CeO2-NC | 28 | 24 | 19 | 2.05 | 1.76 | 335 | 405 |
| Co/CeO2-NR | 72 | 14 | 16 | 2.37 | 1.76 | 318 | 388 |
| Cu/CeO2-NC | 34 | 19 | 52 | 1.50 | 1.34 | 194 | 228 |
| Cu/CeO2-NR | 75 | 12 | 43 | 1.80 | 1.34 | 181 | 217 |
| Sample | % XCO2 | % SCO | % SCH4 | Reaction Rates | |
|---|---|---|---|---|---|
| rs (µmol CO2·m−2·s−1) | rm (µmol CO2·g−1·s−1) | ||||
| CeO2-NR | 21.1 | 88.5 | 11.5 | 0.09 | 7.2 |
| CeO2-NC | 19.3 | 89.8 | 10.2 | 0.18 | 6.6 |
| Cu/CeO2-NR | 55.0 | 97.0 | 3.0 | 0.25 | 18.8 |
| Cu/CeO2-NC | 50.1 | 97.5 | 2.5 | 0.51 | 17.1 |
| Co/CeO2-NR | 84.9 | 5.5 | 94.5 | 0.40 | 28.9 |
| Co/CeO2-NC | 87.7 | 3.7 | 96.3 | 1.07 | 29.9 |
| Sample | T (°C) | % XCO2 | % SCO | % SCH4 | H2:CO2 | %wt. Cu or Co | Ref. |
|---|---|---|---|---|---|---|---|
| Cu-Catalyzed rWGS Reaction | |||||||
| Cu/CeO2-NR | 400 | 19 | 99.6 | 1 | 8.5 | This work | |
| 38 | 99.0 | 4 | |||||
| Fe-Cu/Al2O3 | 400 | 36 | 89 | 4 | 8.2 | [15] | |
| Cu/CeO2 | 400 | 31.3 | 100 | 4 | 13 | [82] | |
| Cu/CeO2-NR | 450 | 49 | N/A | 5 | 5 | [83] | |
| Cu/CeO2 | 300 | ~18 | 100 | 3 | 9 | [60] | |
| Cu-Ni/γ-Al2O3 | 500 | 23.2 | 75.5 | 1 | 15 | [84] | |
| Cu-Fe/SiO2 | 600 | 15 | N/A | 1 | 10 | [85] | |
| Cu/β-Mo2C | 400 | 16 | 97.6 | 2 | 1.3 | [86] | |
| Co-Catalyzed CO2 Methanation | |||||||
| Co/CeO2-NR | 400 | 62.8 | 91.1 | 4 | 7.9 | This work | |
| 84.9 | 94.5 | 9 | |||||
| Co/CeO2 | 400 | 34.9 | 37 | 4 | 10 | [82] | |
| Co/SiO2 | 360 | 44.3 | 86.5 | 4 | 10 | [87] | |
| Co/KIT-6 | 300 | 51 | 98.9 | 4.6 | 20 | [88] | |
| Ni-Co/Ce0.25Zr0.75O2 | 280 | 85 | 98 | 4 | 5 | [89] | |
| Co/Al2O3 | 300 | 38 | 100 | 4 | 15 | [90] | |
| Co/CeO2 | 300 | 97 | ~96 | 9 | 42.3 | [91] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konsolakis, M.; Lykaki, M.; Stefa, S.; Carabineiro, S.A.C.; Varvoutis, G.; Papista, E.; Marnellos, G.E. CO2 Hydrogenation over Nanoceria-Supported Transition Metal Catalysts: Role of Ceria Morphology (Nanorods versus Nanocubes) and Active Phase Nature (Co versus Cu). Nanomaterials 2019, 9, 1739. https://doi.org/10.3390/nano9121739
Konsolakis M, Lykaki M, Stefa S, Carabineiro SAC, Varvoutis G, Papista E, Marnellos GE. CO2 Hydrogenation over Nanoceria-Supported Transition Metal Catalysts: Role of Ceria Morphology (Nanorods versus Nanocubes) and Active Phase Nature (Co versus Cu). Nanomaterials. 2019; 9(12):1739. https://doi.org/10.3390/nano9121739
Chicago/Turabian StyleKonsolakis, Michalis, Maria Lykaki, Sofia Stefa, Sόnia A. C. Carabineiro, Georgios Varvoutis, Eleni Papista, and Georgios E. Marnellos. 2019. "CO2 Hydrogenation over Nanoceria-Supported Transition Metal Catalysts: Role of Ceria Morphology (Nanorods versus Nanocubes) and Active Phase Nature (Co versus Cu)" Nanomaterials 9, no. 12: 1739. https://doi.org/10.3390/nano9121739