The Influence of Initiator Concentration on Selected Properties on Poly-N-Vinylcaprolactam Nanoparticles


Abstract
:
1. Introduction
2. Results
2.1. Synthesis
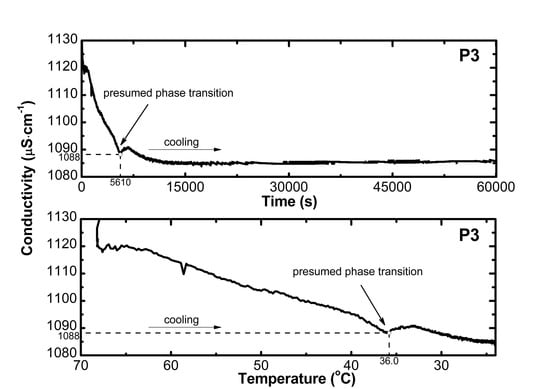
2.2. Conductivity Measurements Analysis
2.3. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy Analysis (ATR-FTIR)
2.4. Nuclear Magnetic Resonance Spectroscopy Analysis (1H NMR)
2.5. Hydrodynamic Diameter (HD)
2.6. Polydispersity Index (PDI)
2.7. Zeta Potential (ZP)
2.8. Thermogravimetric Analysis (TGA)
2.9. Powder X-ray Diffraction Analysis (PXRD)
3. Discussion
3.1. Synthesis
3.2. Conductivity
3.3. ATR-FTIR
3.4. 1H NMR
3.5. HD
3.6. PDI
3.7. ZP
3.8. TG
3.9. PXRD
4. Materials and Methods
4.1. Materials
4.2. Synthesis
4.3. Conductivity Measurements
4.4. Attenuated Total Reflection Fourier Transformed Infrared Spectroscopy Measurements (ATR-FTIR)
4.5. Nuclear Magnetic Resonance Spectroscopy Measurements (1H NMR)
4.6. Hydrodynamic Diameter (HD) and Polydispersity Index (PDI) Measurements
4.7. Zeta Potential (ZP) Measurements
4.8. Thermogravimetric Analysis (TGA)
4.9. Powder X-ray Diffraction Analysis (PXRD)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alvarez-Lorenzo, C.; Concheiro, A. Intelligent Drug Delivery Systems: Polymeric Micelles and Hydrogels. Mini-Rev. Med. Chem. 2008, 8, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Caldorera-Moore, M.; Guimard, N.; Shi, L.; Roy, K. Designer nanoparticles: Incorporating size, shape, and triggered release into nanoscale drug carriers. Expert Opin. Drug Deliv. 2010, 7, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Safari, J.; Zarnegar, Z. Advanced drug delivery systems: Nanotechnology of health design A review. J. Saudi Chem. Soc. 2014, 18, 85–99. [Google Scholar] [CrossRef]
- Honey, P.J.; Rijo, J.; Anju, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef]
- Jun, L.; Bochu, W.; Yazhou, W. Thermo-sensitive polymers for controlled-release drug delivery system. Int. J. Pharmacol. 2006, 2, 513–519. [Google Scholar] [CrossRef]
- Madhusudana Rao, K.; Mallikarjuna, B.; Krishna Rao, K.S.V.; Siraj, S.; Chowdoji Rao, K.; Subha, M.C.S. Novel thermo/pH sensitive nanogels composed from poly(N-vinylcaprolactam) for controlled release of an anticancer drug. Colloids Surf. B 2013, 102, 891–897. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Hoffman, A.S. Stimuli-responsive polymers: Biomedical applications and challenges for clinical translation. Adv. Drug Deliv. Rev. 2013, 65, 10–16. [Google Scholar] [CrossRef]
- Sun, H.; Kabb, C.P.; Sims, M.B.; Sumerlin, B.S. Architecture-transformable polymers: Reshaping the future of stimuli-responsive polymers. Prog. Polym. Sci. 2019, 89, 61–75. [Google Scholar] [CrossRef]
- Yang, L.; Sun, H.; Liu, Y.; Hou, W.; Yang, Y.; Cai, R.; Cui, C.; Zhang, P.; Pan, X.; Li, X.; et al. Self-Assembled Aptamer-Grafted Hyperbranched Polymer Nanocarrier for Targeted and Photoresponsive Drug Delivery. Angew. Chem. Int. Ed. 2018, 57, 17048–17052. [Google Scholar] [CrossRef]
- Sun, H.; Choi, W.; Zang, N.; Battistella, C.; Thompson, M.; Cao, W.; Zhou, X.; Forman, C.; Gianneschi, N.C. Bioactive Peptide Brush Polymers via Photoinduced Reversible-Deactivation Radical Polymerization. Angew. Chem. Int. Edit. 2019. [Google Scholar] [CrossRef]
- Gil, E.S.; Hudson, S.M. Stimuli-responsive polymers and their bioconjugates. Prog. Polym. Sci. 2004, 29, 1173–1222. [Google Scholar] [CrossRef]
- Crespy, D.; Golosova, A.; Makhaeva, E.; Khokhlov, A.R.; Fortunato, G.; Rossi, R. Synthesis and characterization of temperature-responsive copolymers based on N-vinylcaprolactam and their grafting on fibres. Polym. Int. 2009, 58, 1326–1334. [Google Scholar] [CrossRef]
- Medeiros, S.F.; Barboza, J.C.S.; Giudici, R.; Santos, A.M. Thermally-sensitive and Biocompatible Poly(N-vinylcaprolactam): A Kinetic Study of Free Radical Polymerization in Ethanol. J. Macromol. Sci. 2013, 50, 763–773. [Google Scholar] [CrossRef]
- Kozanoglu, S.; Ozdemir, T.; Usanmaz, A. Polymerization of N-Vinylcaprolactam and Characterization of Poly(N-Vinylcaprolactam). J. Macromol. Sci. 2011, 48, 467–477. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Licea-Claverie, A. Poly(N-vinylcaprolactam), a comprehensive review on a thermoresponsive polymer becoming popular. Prog. Polym. Sci. 2016, 53, 1–51. [Google Scholar] [CrossRef]
- Ramos, J.; Imaz, A.; Forcada, J. Temperature-sensitive nanogels: Poly(N-vinylcaprolactam) versus poly(N-isopropylacrylamide). Polym. Chem. 2012, 3, 852–856. [Google Scholar] [CrossRef]
- Chihacheva, I.P.; Timaeva, O.I.; Kuz’micheva, G.M.; Dorohov, A.V.; Lobanova, N.A.; Amarantov, S.V.; Podbel’skiy, V.V.; Serousov, V.E.; Sadovskaya, N.V. Specific Physical and Chemical Properties of Two Modifications of Poly(N-vinylcaprolactam). Crystallogr. Rep. 2016, 61, 421–427. [Google Scholar] [CrossRef]
- Umapathi, R.; Venkatesu, P. Assessing the efficiency of imidazolium-based ionic liquids on the phase behavior of a synthetic biomedical thermoresponsive polymer. J. Colloid Interface Sci. 2018, 511, 174–183. [Google Scholar] [CrossRef]
- Richard, A. Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra, 1st ed.; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Gola, A.; Knysak, T.; Musiał, W. The Influence of Anionic Initiator on the Selected Properties of Poly-N-Isopropyl Acrylamide Evaluated for Controlled Drug Delivery. Molecules 2017, 22, 23. [Google Scholar] [CrossRef]
- Gola, A.; Sacharczuk, M.; Musiał, W. Synthesis of AMPSA polymeric derivatives monitored by electrical conductivity and evaluation of thermosensitive properties of resulting microspheres. Molecules 2019, 24, 1164. [Google Scholar] [CrossRef] [PubMed]
- SDBSWeb. National Institute of Advanced Industrial Science and Technology. Available online: http://sdbs.db.aist.go.jp (accessed on 4 November 2019).
- Wu, X.S.; Hoffman, A.S.; Yager, P. Synthesis and characterization of thermally reversible macroporous Poly (N-isopropylacrylamide) hydrogels. J. Polym. Sci. Pol. Chem. 1992, 30, 2121–2129. [Google Scholar] [CrossRef]
- Aseyev, V.; Hietala, S.; Laukkanen, A.; Nuopponen, M.; Confortini, O.; Du Prez, F.E.; Tenhu, H. Mesoglobules of thermoresponsive polymers in dilute aqueous solutions above the LCST. Polymer 2005, 46, 7118–7131. [Google Scholar] [CrossRef]
- Musiał, W.; Pluta, J.; Michálek, J. Thermosensitive microgels of poly-N-isopropylacrylamide for drug carriers—practical approach to synthesis. Acta Pol. Pharm. 2015, 72, 409–422. [Google Scholar]
- Gasztych, M.; Kotowska, A.; Musiał, W. Application of polymerization activator in the course of synthesis of N-isopropylacrylamide derivatives for thermally triggered release of naproxen sodium. Materials 2018, 11, 261. [Google Scholar] [CrossRef]
- Tan, S.; Lu, Z.; Zhao, J.; Zhang, J.; Wu, M.; Wu, Q.; Yang, J. Synthesis and multi-responsiveness of poly(N-vinylcaprolactam-co-acrylic acid) core-shell microgels via miniemulsion polymerization. Polym. Chem. 2016, 7, 4106–4111. [Google Scholar] [CrossRef]
- Eslami, H.; Bahri, K.; Müller-Plathe, F. Solid-Liquid and Solid-Solid Phase Diagrams of Self-Assembled Triblock Janus Nanoparticles from Solution. J. Phys. Chem. C 2018, 38, 9235–9244. [Google Scholar] [CrossRef]
- Eslami, H.; Khanjari, N.; Müller-Plathe, F. Self-Assembly Mechanisms of Triblock Janus Particles. J. Chem. Theory Comput. 2019, 15, 1345–1354. [Google Scholar] [CrossRef]
- Matthews, B.A.; Rhodes, C.T. Use of the Derjaguin, Landau, Verwey and Overbeek theory to interpret pharmaceutical suspension stability. J. Pharm. Sci. 1970, 59, 521–525. [Google Scholar] [CrossRef]
- Horák, D.; Kryŝtủfek, M.; Spĕváček. Effect of Reaction Parameters on the Dispersion Polymerization of 1-Vinyl-2-Pyrrolidone. J. Polym. Sci. Polym. Chem. 2000, 38, 653–663. [Google Scholar] [CrossRef]
- Ober, C.K.; Hair, M.L. The Effect of Temperature and Initiator Levels on the Dispersion Polymerization of Polystyrene. J. Polym. Sci. Polym. Chem. 1987, 25, 1395–1407. [Google Scholar] [CrossRef]
- Bai, F.; Yang, X.; Huang, W. Synthesis of Narrow or Monodisperse Poly(divinylbenzene) Microspheres by Distillation-Precipitation Polymerization. Macromolecules 2004, 37, 9746–9752. [Google Scholar] [CrossRef]
- Liu, X.X.; Dig, X.B.; Zheng, Z.H.; Peng, Y.X.; Long, X.P.; Wang, X.C.; Chan, A.S.C.; Yip, C.W. Synthesis of Novel Magnetic Polymer Microspheres with Amphiphilic Structure. J. Appl. Polym. Sci. 2003, 90, 1879–1884. [Google Scholar] [CrossRef]
- FDA—Liposome Drug Products; Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; Labeling Documentation. Guidance for Industry; April 2018 Pharmaceutical Quality/CMC; U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER): Silver Spring, MD, USA, 2018.
- Musiał, W.; Michálek, J. The influence of low process temperature on the hydrodynamic radius of polyNIPAM-co-PEG thermosensitive nanoparticles presumed as drug carriers for bioactive proteins. Acta Pol. Pharm. 2015, 72, 161–169. [Google Scholar]
- International Standard. ISO 13321:1996. Methods for Determination of Particle Size Distribution. Photon Correlation Spectroscopy; International Organization for Standardization (ISO): Geneva, Switzerland, 1997; pp. 3406–3408. [Google Scholar]
- International Standard. ISO22412 Particle Size Analysis–Dynamic Light Scattering; International Organisation for Standardisation (ISO): Geneva, Switzerland, 2008. [Google Scholar]
- Worldwide, M.I. Dynamic Light Scattering, Common Terms Defined; Inform White Paper; Malvern Instruments Limited: Malvern, UK, 2011; pp. 1–6. [Google Scholar]
Sample Availability: Samples of the compounds P1-P5 are available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Polymer Nanoparticles System | Monomer (mol) | Cationic Initiator (mol) |
|---|---|---|
| NVCL | AMPA | |
| P1 | 2.184 × 10−2 | 16.165 × 10−3 |
| P2 | 2.155 × 10−2 | 10.777 × 10−3 |
| P3 | 2.187 × 10−2 | 5.391 × 10−3 |
| P4 | 2.160 × 10−2 | 2.695 × 10−3 |
| P5 | 2.163 × 10−2 | 1.347 × 10−3 |
| Type of Polymer Nanoparticle System | Tm1 (°C) | Rate of Mass Loss 1 (% min−1) | Tm2 (°C) | Rate of Mass Loss 2 (% min−1) | TOnset (°C) | TEndset (°C) | Res (%) | T0.5wt% (°C) |
|---|---|---|---|---|---|---|---|---|
| P1 | 51.5 | 0.92 | 437.7 | 18.63 | 410.1 | 457.2 | 5.03 | 29.7 |
| P2 | 52.2 | 0.90 | 437.3 | 18.41 | 408.8 | 457.1 | 3.68 | 29.5 |
| P3 | 48.1 | 0.68 | 436.5 | 18.36 | 404.8 | 457.1 | 4.22 | 30.2 |
| P4 | 53.7 | 0.71 | 436.0 | 18.06 | 404.9 | 456.9 | 3.62 | 30.4 |
| P5 | 52.4 | 0.75 | 437.6 | 17.99 | 402.9 | 457.1 | 4.11 | 29.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gola, A.; Niżniowska, A.; Musiał, W. The Influence of Initiator Concentration on Selected Properties on Poly-N-Vinylcaprolactam Nanoparticles. Nanomaterials 2019, 9, 1577. https://doi.org/10.3390/nano9111577
Gola A, Niżniowska A, Musiał W. The Influence of Initiator Concentration on Selected Properties on Poly-N-Vinylcaprolactam Nanoparticles. Nanomaterials. 2019; 9(11):1577. https://doi.org/10.3390/nano9111577
Chicago/Turabian StyleGola, Agnieszka, Aleksandra Niżniowska, and Witold Musiał. 2019. "The Influence of Initiator Concentration on Selected Properties on Poly-N-Vinylcaprolactam Nanoparticles" Nanomaterials 9, no. 11: 1577. https://doi.org/10.3390/nano9111577