A New Route for Low Pressure and Temperature CWAO: A PtRu/MoS2_Hyper-Crosslinked Nanocomposite



 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Nanoparticles
2.3. Synthesis of the Nanocomposite
2.4. Catalytic Tests
2.5. Characterization Methods
3. Results
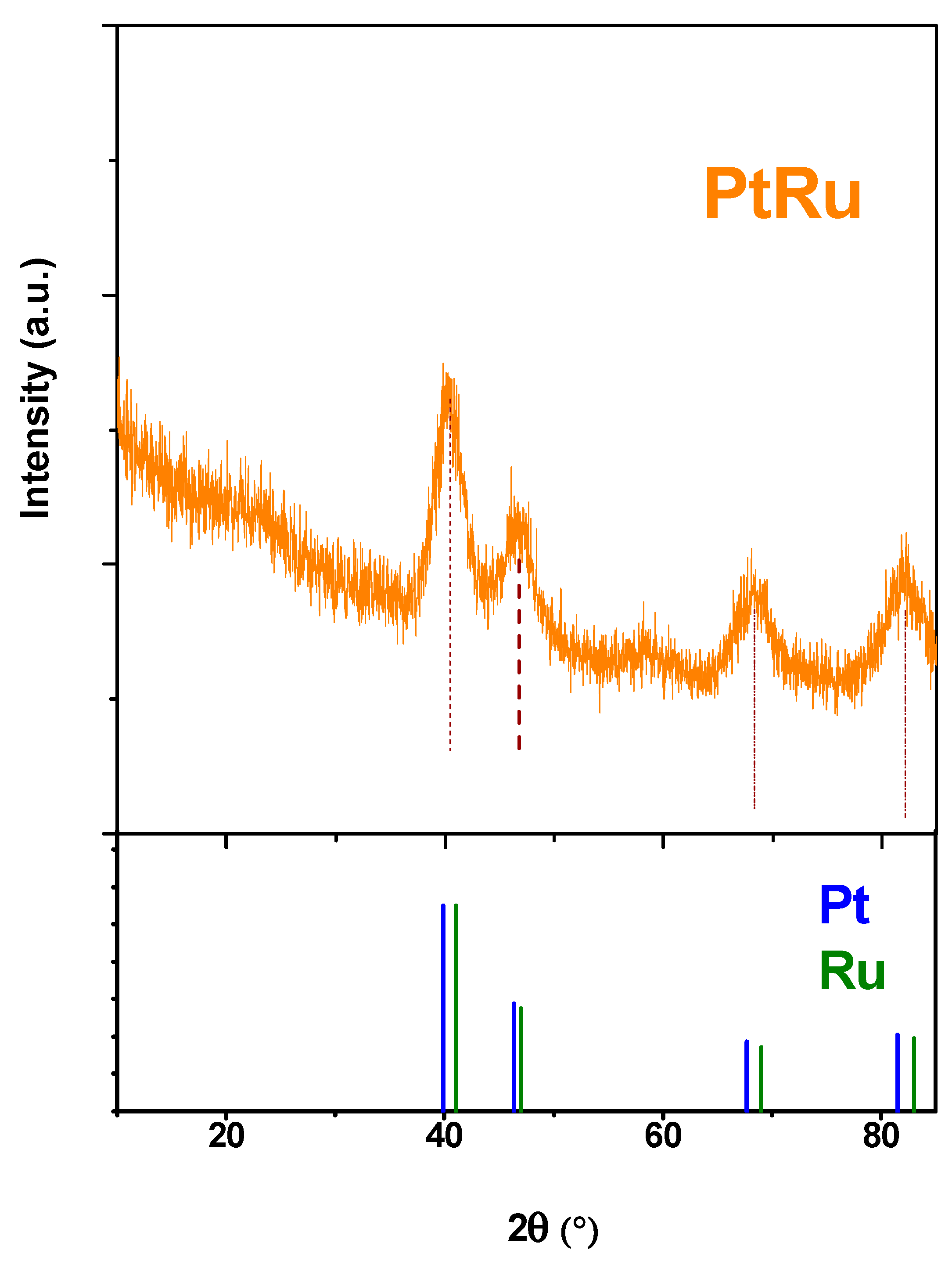
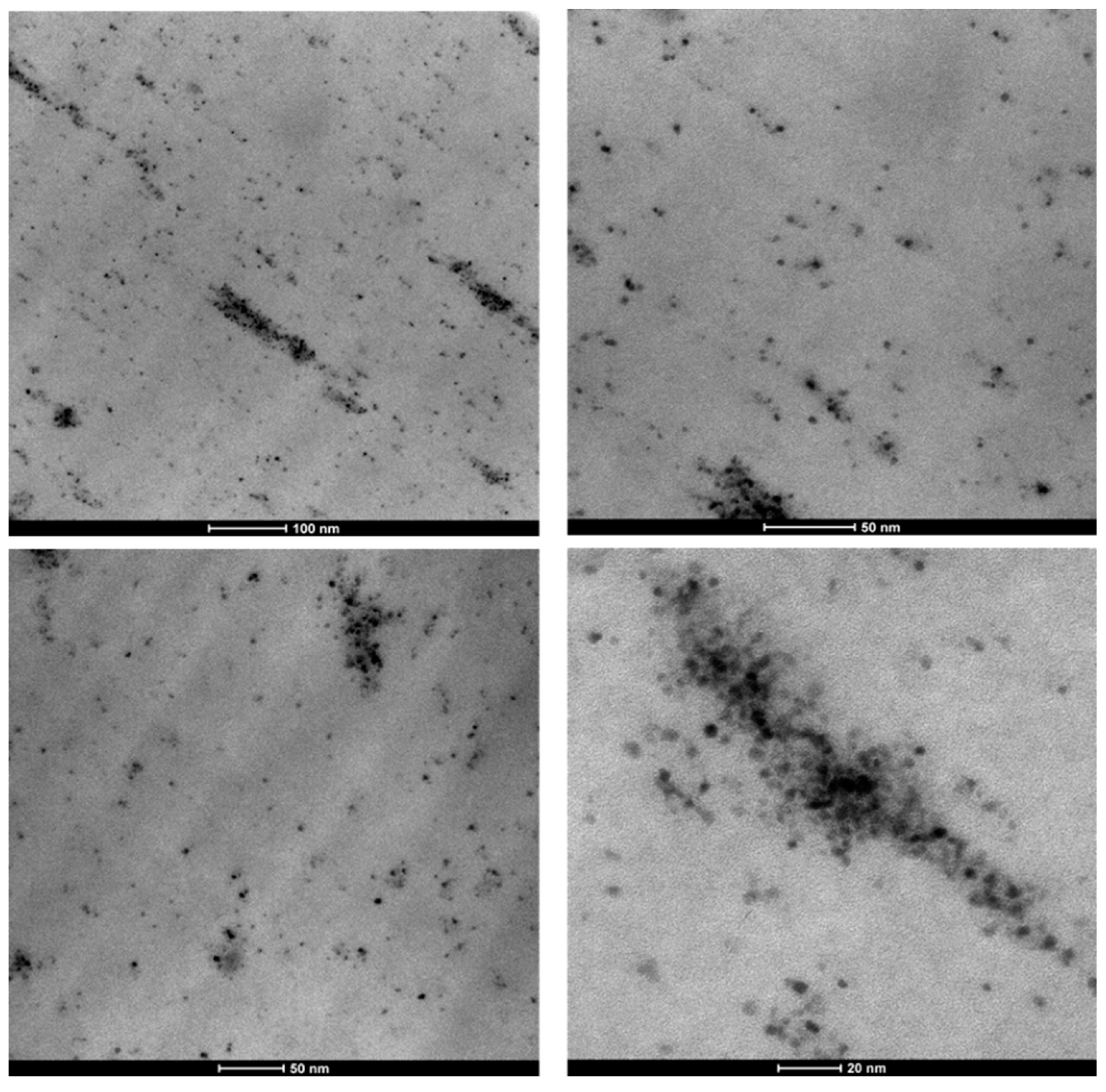
3.1. Nanocatalyst Characterization
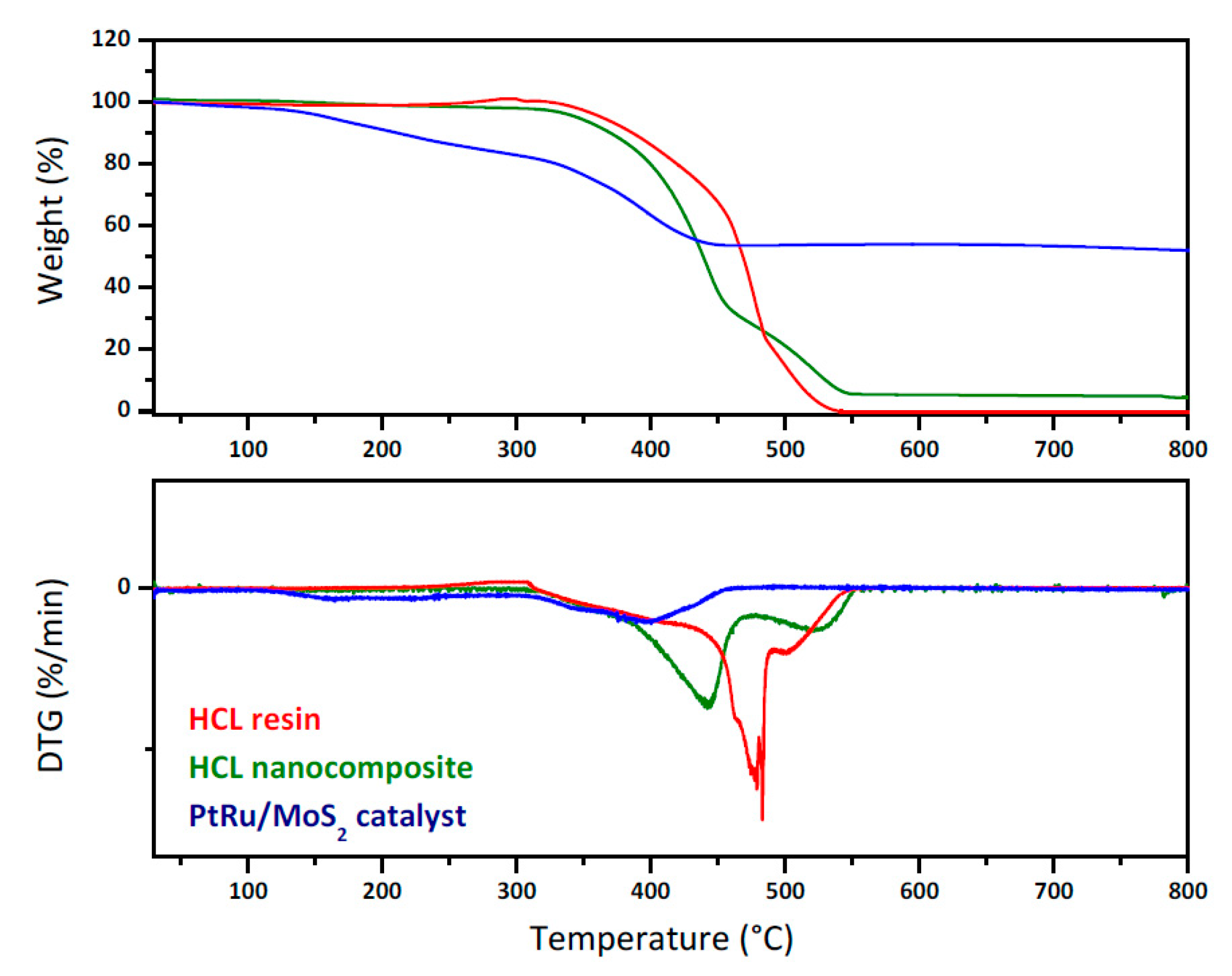
3.2. Resin Characterization
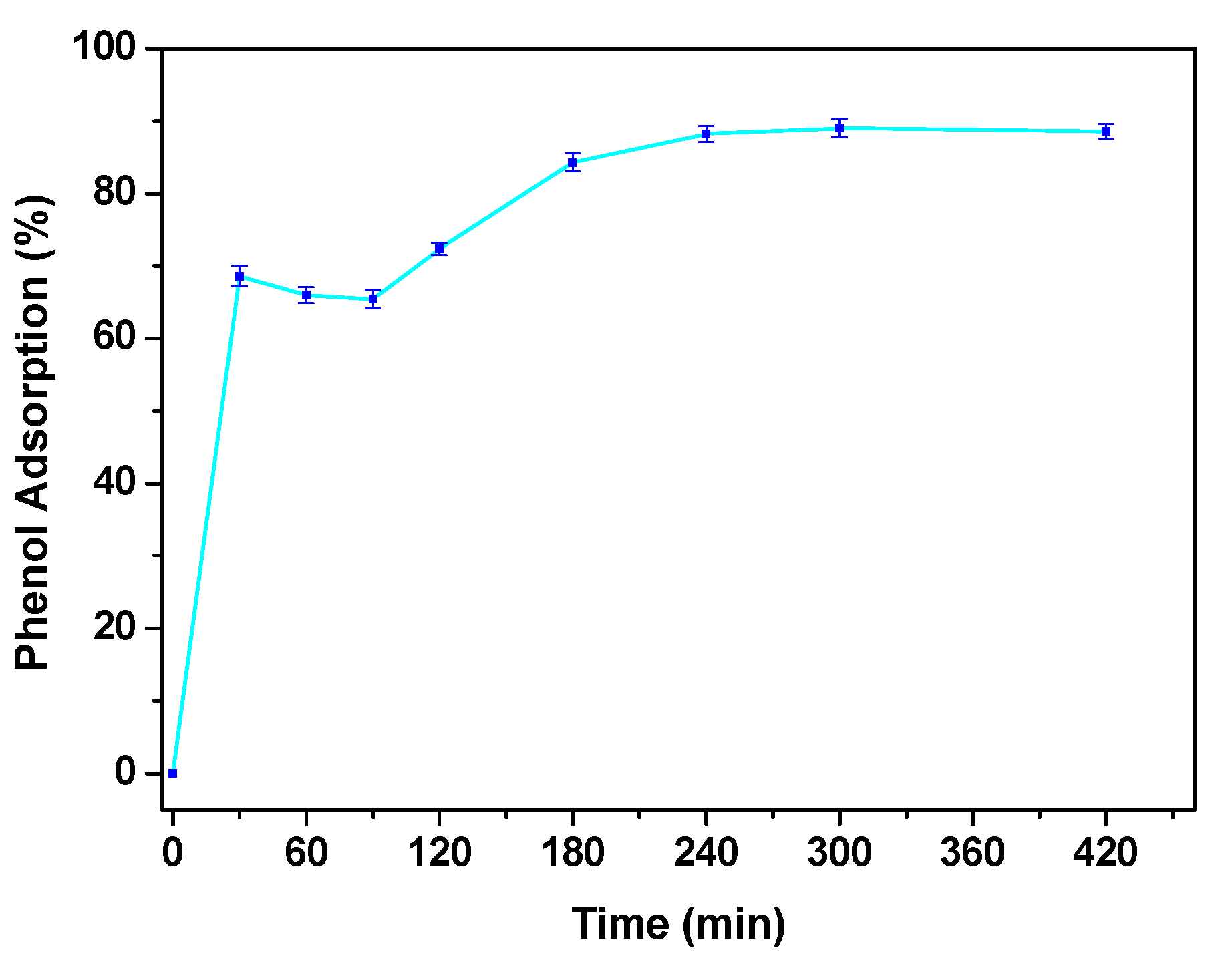
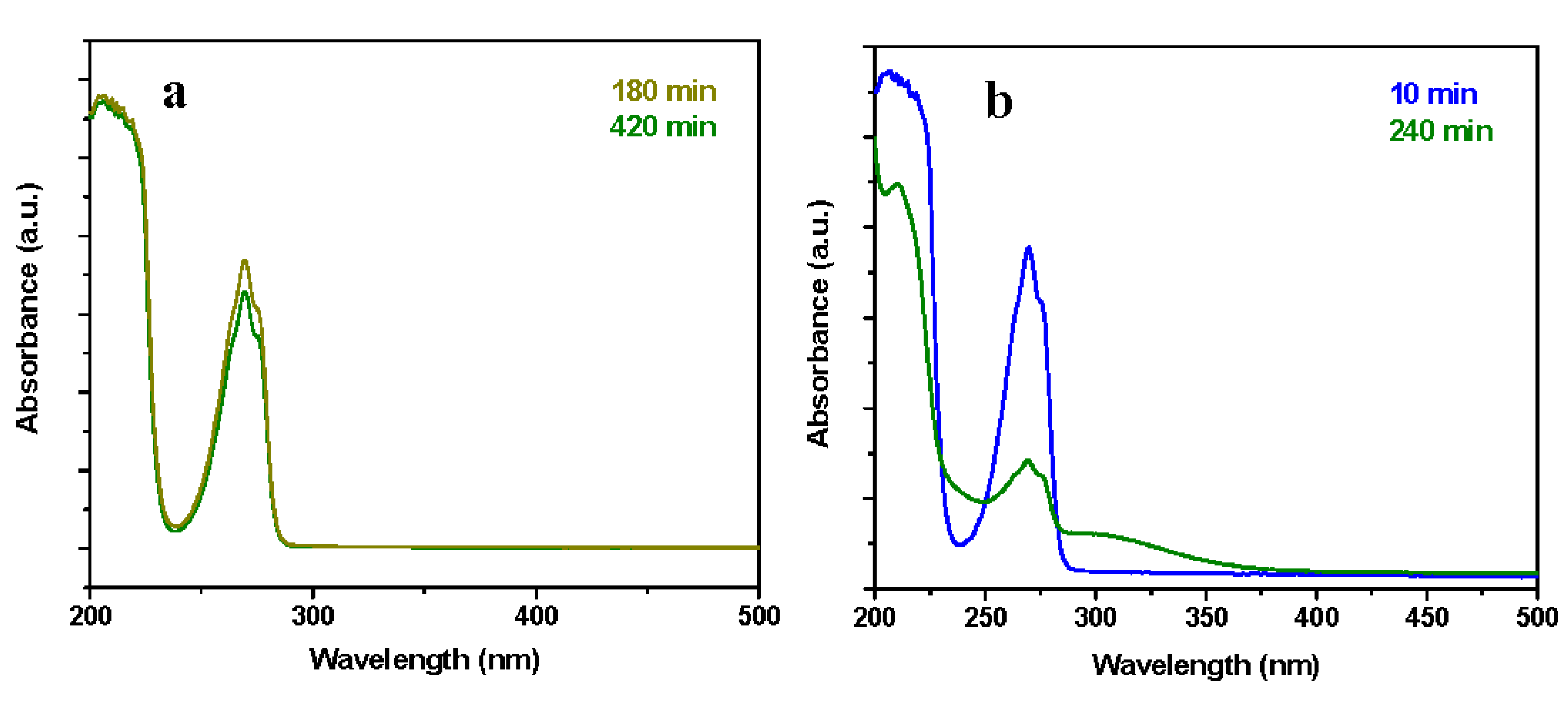
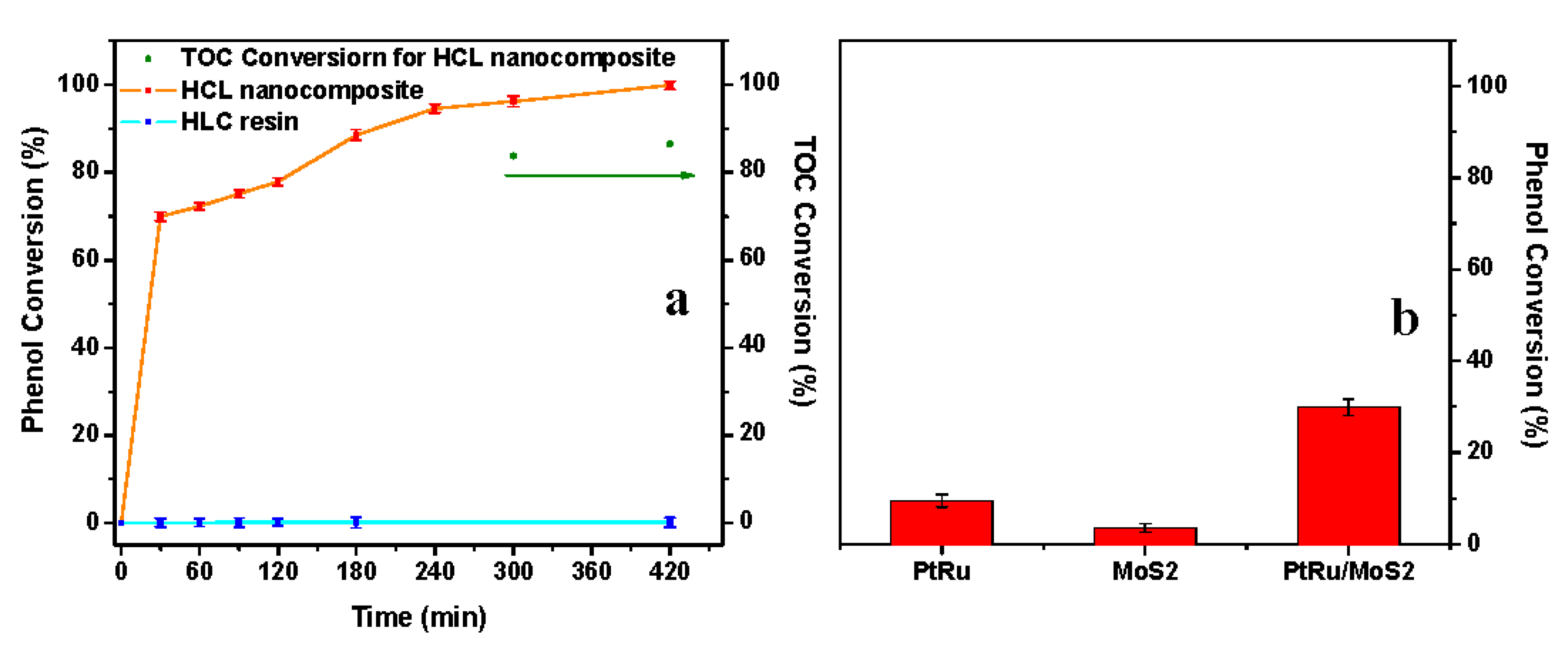
3.3. Catalyst Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruce, R.M.; Santodonato, J.; Neal, M.W. Summary Review of the Health Effects Associated with Phenol. Toxicol. Ind. Health 1987, 3, 535–568. [Google Scholar] [CrossRef] [PubMed]
- Tisler, T.; Zagorc-Končan, J. Comparative assessment of toxicity of phenol, formaldehyde, and industrial wastewater to aquatic organisms. Water Air Soil Pollut. 1997, 97, 315–322. [Google Scholar] [CrossRef]
- Saha, N.C.; Bhunia, F.; Kaviraj, A. Toxicity of Phenol to Fish and Aquatic Ecosystems. Bull. Environ. Contam. Toxicol. 1999, 63, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, P.; Gloyna, E.F. Generalized kinetic model for wet oxidation of organic compounds. AIChE J. 1991, 37, 1687–1697. [Google Scholar] [CrossRef]
- Eftaxias, A.; Font, J.; Fortuny, A.; Giralt, J.; Fabregat, A.; Stuber, F. Kinetic modelling of catalytic wet air oxidation of phenol by simulated annealing. Appl. Catal. B Environ. 2001, 33, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Rivas, F.; Kolaczkowski, S.; Beltran, F.J.; McLurgh, D. Development of a model for the wet air oxidation of phenol based on a free radical mechanism. Chem. Eng. Sci. 1998, 53, 2575–2586. [Google Scholar] [CrossRef]
- Santos, A.; Yustos, P.; Quintanilla, A.; García-Ochoa, F.; Casas, J.A.; Rodríguez, J.J. Evolution of Toxicity upon Wet Catalytic Oxidation of Phenol. Environ. Sci. Technol. 2004, 38, 133–138. [Google Scholar] [CrossRef]
- Danis, T.G.; Albanis, T.A.; Petrakis, D.E.; Pomonis, P.J. Removal of chlorinated phenols from aqueous solutions by adsorption on alumina pillared clays and mesoporous alumina aluminum phosphates. Water Res. 1998, 32, 295–302. [Google Scholar] [CrossRef]
- Sincero, A.P.; Sincero, G.A. Physical-Chemical Treatment of Water and Wastewater; CRC Press: Boca Raton, FL, USA, 2003; ISBN 9781587161247. [Google Scholar]
- Kolaczkowski, S.; Plucinski, P.; Beltran, F.J.; Rivas, F.; McLurgh, D.; Rivas, J. Wet air oxidation: A review of process technologies and aspects in reactor design. Chem. Eng. J. 1999, 73, 143–160. [Google Scholar] [CrossRef]
- Mishra, V.S.; Mahajani, V.V.; Joshi, J.B. Wet Air Oxidation. Ind. Eng. Chem. Res. 1995, 34, 2–48. [Google Scholar] [CrossRef]
- Luck, F. Wet air oxidation: Past, present and future. Catal. Today 1999, 53, 81–91. [Google Scholar] [CrossRef]
- Pintar, A.; Besson, M.; Gallezot, P. Catalytic wet air oxidation of Kraft bleaching plant effluents in the presence of titania and zirconia supported ruthenium. Appl. Catal. B Environ. 2001, 30, 123–139. [Google Scholar] [CrossRef]
- Luck, F. A review of industrial catalytic wet air oxidation processes. Catal. Today 1996, 27, 195–202. [Google Scholar] [CrossRef]
- Liotta, L.; Gruttadauria, M.; Di Carlo, G.; Perrini, G.; Librando, V.; Liotta, L. Heterogeneous catalytic degradation of phenolic substrates: Catalysts activity. J. Hazard. Mater. 2009, 162, 588–606. [Google Scholar] [CrossRef]
- Villegas, L.G.C.; Mashhadi, N.; Chen, M.; Mukherjee, D.; Taylor, K.E.; Biswas, N. A Short Review of Techniques for Phenol Removal from Wastewater. Curr. Pollut. Rep. 2016, 2, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Levec, J.; Pintar, A. Catalytic wet-air oxidation processes: A review. Catal. Today 2007, 124, 172–184. [Google Scholar] [CrossRef]
- Arena, F.; Di Chio, R.; Gumina, B.; Spadaro, L.; Trunfio, G. Recent advances on wet air oxidation catalysts for treatment of industrial wastewaters. Inorg. Chim. Acta 2015, 431, 101–109. [Google Scholar] [CrossRef]
- Kim, K.-H.; Ihm, S.-K. Heterogeneous catalytic wet air oxidation of refractory organic pollutants in industrial wastewaters: A review. J. Hazard. Mater. 2011, 186, 16–34. [Google Scholar] [CrossRef]
- Sulman, E.M.; Matveeva, V.G.; Doluda, V.Y.; Sidorov, A.I.; Lakina, N.V.; Bykov, A.V.; Sulman, M.G.; Valetsky, P.M.; Kustov, L.M.; Tkachenko, O.P. Efficient polymer-based nanocatalysts with enhanced catalytic performance in wet air oxidation of phenol. Appl. Catal. B Environ. 2010, 94, 200–210. [Google Scholar] [CrossRef]
- Doluda, V.Y.; Sulman, E.M.; Matveeva, V.G.; Sulman, M.G.; Bykov, A.V.; Lakina, N.V.; Sidorov, A.I.; Valetsky, P.M.; Bronstein, L.M.; Sulman, M. Phenol Catalytic Wet Air Oxidation Over Ru Nanoparticles Formed in Hypercrosslinked Polystyrene. Top. Catal. 2013, 56, 688–695. [Google Scholar] [CrossRef]
- Doluda, V.; Sulman, E.; Matveeva, V.; Sulman, M.; Lakina, N.; Sidorov, A.; Valetsky, P.; Bronstein, L.; Sulman, M. Kinetics of phenol oxidation over hypercrosslinked polystyrene impregnated with Pt nanoparticles. Chem. Eng. J. 2007, 134, 256–261. [Google Scholar] [CrossRef]
- Nousir, S.; Keav, S.; Barbier, J.; Bensitel, M.; Brahmi, R.; Duprez, D. Deactivation phenomena during catalytic wet air oxidation (CWAO) of phenol over platinum catalysts supported on ceria and ceria–zirconia mixed oxides. Appl. Catal. B Environ. 2008, 84, 723–731. [Google Scholar] [CrossRef]
- Taboada, C.D.; Batista, J.; Pintar, A.; Levec, J. Preparation, characterization and catalytic properties of carbon nanofiber-supported Pt, Pd, Ru monometallic particles in aqueous-phase reactions. Appl. Catal. B Environ. 2009, 89, 375–382. [Google Scholar] [CrossRef]
- Masende, Z.P.G.; Kuster, B.F.M.; Ptasinski, K.J.; Janssen, F.J.J.G.; Katima, J.H.Y.; Schouten, J.C. Support and dispersion effects on activity of platinum catalysts during wet oxidation of organic wastes. Top. Catal. 2005, 33, 87–99. [Google Scholar] [CrossRef]
- Ayusheev, A.B.; Taran, O.P.; Seryak, I.A.; Podyacheva, O.Y.; Descorme, C.; Besson, M.; Kibis, L.S.; Boronin, A.I.; Romanenko, A.I.; Ismagilov, Z.R.; et al. Ruthenium nanoparticles supported on nitrogen-doped carbon nanofibers for the catalytic wet air oxidation of phenol. Appl. Catal. B Environ. 2014, 146, 177–185. [Google Scholar] [CrossRef]
- Barbier, J.; Oliviero, L.; Renard, B.; Duprez, D. Role of ceria-supported noble metal catalysts (Ru, Pd, Pt) in wet air oxidation of nitrogen and oxygen containing compounds. Top. Catal. 2005, 33, 77–86. [Google Scholar] [CrossRef]
- Oliviero, L.; Barbier, J.; Duprez, D.; Guerrero-Ruiz, A.; Bachiller-Baeza, B.; Rodríguez-Ramos, I. Catalytic wet air oxidation of phenol and acrylic acid over Ru/C and Ru–CeO2/C catalysts. Appl. Catal. B Environ. 2000, 25, 267–275. [Google Scholar] [CrossRef]
- Cao, S.; Chen, G.; Hu, X.; Yue, P.L. Catalytic wet air oxidation of wastewater containing ammonia and phenol over activated carbon supported Pt catalysts. Catal. Today 2003, 88, 37–47. [Google Scholar] [CrossRef]
- Gallezot, P. Catalytic wet-air oxidation of carboxylic acids on carbon-supported platinum catalysts. Appl. Catal. B: Environ. 1996, 9, L11–L17. [Google Scholar] [CrossRef]
- Fortuny, A.; Bengoa, C.; Font, J.; Fabregat, A. Bimetallic catalysts for continuous catalytic wet air oxidation of phenol. J. Hazard. Mater. 1999, 64, 181–193. [Google Scholar] [CrossRef]
- Hočevar, S.; Krašovec, U.O.; Orel, B.; Arico’, A.S.; Kim, H. CWO of phenol on two differently prepared CuO–CeO2 catalysts. Appl. Catal. B Environ. 2000, 28, 113–125. [Google Scholar] [CrossRef]
- Chen, H.; Sayari, A.; Adnot, A.; Larachi, F. Composition–activity effects of Mn–Ce–O composites on phenol catalytic wet oxidation. Appl. Catal. B Environ. 2001, 32, 195–204. [Google Scholar] [CrossRef]
- Kim, K.-H.; Kim, J.-R.; Ihm, S.-K. Wet oxidation of phenol over transition metal oxide catalysts supported on Ce0.65Zr0.35O2 prepared by continuous hydrothermal synthesis in supercritical water. J. Hazard. Mater. 2009, 167, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Chen, C.L.; Chang, D.J.; Chen, C.C. Catalytic wet air oxidation of phenol by various CeO2 catalysts. Water Res. 2002, 36, 3009–3014. [Google Scholar] [CrossRef]
- Lin, S.S.; Chang, D.J.; Wang, C.-H.; Chen, C.C. Catalytic wet air oxidation of phenol by CeO2 catalyst—Effect of reaction conditions. Water Res. 2003, 37, 793–800. [Google Scholar] [CrossRef]
- Chen, I.-P.; Lin, S.-S.; Wang, C.-H.; Chang, S.-H. CWAO of phenol using CeO2/γ-Al2O3 with promoter—Effectiveness of promoter addition and catalyst regeneration. Chemosphere 2007, 66, 172–178. [Google Scholar] [CrossRef]
- Guo, J.; Al-Dahhan, M. Kinetics of Wet Air Oxidation of Phenol over a Novel Catalyst. Ind. Eng. Chem. Res. 2003, 42, 5473–5481. [Google Scholar] [CrossRef]
- Wang, J.; Fu, W.; He, X.; Yang, S.; Zhu, W. Catalytic wet air oxidation of phenol with functionalized carbon materials as catalysts: Reaction mechanism and pathway. J. Environ. Sci. 2014, 26, 1741–1749. [Google Scholar] [CrossRef]
- Cao, Y.; Li, B.; Zhong, G.; Li, Y.; Wang, H.; Yu, H.; Peng, F. Catalytic wet air oxidation of phenol over carbon nanotubes: Synergistic effect of carboxyl groups and edge carbons. Carbon 2018, 133, 464–473. [Google Scholar] [CrossRef]
- Castillejos, E.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Catalytic Removal of Water-Solved Aromatic Compounds by Carbon-Based Materials. In Novel Carbon Adsorbents; Elsevier: Amsterdam BV, The Netherlands, 2012; pp. 499–520. [Google Scholar]
- Yang, S.; Zhu, W.; Li, X.; Wang, J.; Zhou, Y. Multi-walled carbon nanotubes (MWNTs) as an efficient catalyst for catalytic wet air oxidation of phenol. Catal. Commun. 2007, 8, 2059–2063. [Google Scholar] [CrossRef]
- Gomes, H.T.; Machado, B.F.; Ribeiro, A.; Moreira, I.; Rosário, M.; Silva, A.M.; Figueiredo, J.L.; Faria, J.L. Catalytic properties of carbon materials for wet oxidation of aniline. J. Hazard. Mater. 2008, 159, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Vaschetto, E.G.; Sicardi, M.I.; Elías, V.R.; Ferrero, G.O.; Carraro, P.M.; Casuscelli, S.G.; Eimer, G.A. Metal modified silica for catalytic wet air oxidation (CWAO) of glyphosate under atmospheric conditions. Adsorption 2019, 25, 1299–1306. [Google Scholar] [CrossRef]
- Zhang, Z.; Ruoyan, Y.; Yanshan, G.; Yufei, Z.; Junyang, W.; Liang, H.; Jiang, G.; Tuantuan, Z.; Peng, L.; Zhanhu, G.; et al. Novel Na2Mo4O13/a-MoO3 hybrid material as highly efficient CWAO catalyst for dye degradation at ambient conditions. Sci. Rep. 2014, 4, 6797. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Tan, B. Hypercrosslinked porous polymer materials: Design, synthesis, and applications. Chem. Soc. Rev. 2017, 46, 3322–3356. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, R.; Avolio, R.; Cocca, M.; Gentile, G.; Errico, M.E.; Avella, M.; Carfagna, C.; Ambrogi, V. Synthesis and adsorption study of hyper-crosslinked styrene-based nanocomposites containing multi-walled carbon nanotubes. RSC Adv. 2017, 7, 6865–6874. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, R.; Gentile, G.; Avella, M.; Carfagna, C.; Ambrogi, V. Microporous Hyper-Crosslinked Polystyrenes and Nanocomposites with High Adsorption Properties: A Review. Polymers 2017, 9, 651. [Google Scholar] [CrossRef]
- Li, A.; Zhang, Q.; Zhang, G.; Chen, J.; Fei, Z.; Liu, F. Adsorption of phenolic compounds from aqueous solutions by a water-compatible hypercrosslinked polymeric adsorbent. Chemosphere 2002, 47, 981–989. [Google Scholar] [CrossRef]
- Altavilla, C.; Ciambelli, P.; Sarno, M. “one-pot” Synthesis of 2d, 1d, e 0d nano Crystals of Tungsten and Molybdenum Chalcogenides (WS2, MoS2) Functionalized with Long Chain Amine and/or Carboxylic Acid and/or Thiol. European Patent Office EP2621859B1, 1 October 2011. [Google Scholar]
- Altavilla, C.; Sarno, M.; Ciambelli, P. A Novel Wet Chemistry Approach for the Synthesis of Hybrid 2D Free-Floating Single or Multilayer Nanosheets of MS2@oleylamine (M=Mo, W). Chem. Mater. 2011, 23, 3879–3885. [Google Scholar] [CrossRef]
- Castaldo, R.; Avolio, R.; Cocca, M.; Gentile, G.; Errico, M.E.; Avella, M.; Carfagna, C.; Ambrogi, V. A Versatile Synthetic Approach toward Hyper-Cross-Linked Styrene-Based Polymers and Nanocomposites. Macromolecules 2017, 50, 4132–4143. [Google Scholar] [CrossRef] [Green Version]
- Sarno, M.; Iuliano, M.; Polichetti, M.; Ciambelli, P. High activity and selectivity immobilized lipase on Fe3O4 nanoparticles for banana flavour synthesis. Process. Biochem. 2017, 56, 98–108. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H.; Robinson, D.B.; Raoux, S.; Rice, P.M.; Wang, S.X.; Li, G. Monodisperse MFe2O4(M = Fe, Co, Mn) Nanoparticles. J. Am. Chem. Soc. 2004, 126, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Sarno, M.; Iuliano, M. Highly active and stable Fe3O4/Au nanoparticles supporting lipase catalyst for biodiesel production from waste tomato. Appl. Surf. Sci. 2019, 474, 135–146. [Google Scholar] [CrossRef]
- Tang, Z.; Li, S.; Yang, W.; Yu, X. Hypercrosslinked porous poly(styrene-co-divinylbenzene) resin: A promising nanostructure-incubator for hydrogen storage. J. Mater. Chem. 2012, 22, 12752. [Google Scholar] [CrossRef]
- Yang, Y.; Tan, B.; Wood, C.D. Solution-processable hypercrosslinked polymers by low cost strategies: A promising platform for gas storage and separation. J. Mater. Chem. A 2016, 4, 15072–15080. [Google Scholar] [CrossRef]
- Sarno, M.; Troisi, A. Supercapacitors Based on High Surface Area MoS2 and MoS2–Fe3O4 Nanostructures Supported on Physical Exfoliated Graphite. J. Nanosci. Nanotechnol. 2017, 17, 3735–3743. [Google Scholar] [CrossRef]
- Sarno, M.; Ponticorvo, E.; Cirillo, C. High surface area monodispersed Fe3O4 nanoparticles alone and on physical exfoliated graphite for improved supercapacitors. J. Phys. Chem. Solids 2016, 99, 138–147. [Google Scholar] [CrossRef]
- Sarno, M.; Ponticorvo, E.; Scarpa, D. PtRh and PtRh/MoS2 nano-electrocatalysts for methanol oxidation and hydrogen evolution reactions. Chem. Eng. J. 2018, in press. [Google Scholar] [CrossRef]
- Sarno, M.; Ponticorvo, E. Effect of the amount of nickel sulphide, molybdenum disulphide and carbon nanosupport on a Tafel slope and overpotential optimization. Nanotechnology 2017, 28, 214003. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodríguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, R.; Ambrogi, V.; Avolio, R.; Cocca, M.; Gentile, G.; Errico, M.E.; Avella, M. Functional hyper-crosslinked resins with tailored adsorption properties for environmental applications. Chem. Eng. J. 2019, 362, 497–503. [Google Scholar] [CrossRef]
- Dauengauer, S.A.; Utkina, O.G.; Popova, G.S.; Sazanov, Y.N. Investigation of thermal degradation of polystyrene with the aid of thermal analysis. J. Therm. Anal. Calorim. 1987, 32, 311–314. [Google Scholar] [CrossRef]
- Emerson, E. The condensation of aminoantipyrine. Ii. a new color test for phenolic compounds. J. Org. Chem. 1943, 8, 417–428. [Google Scholar] [CrossRef]
- Klinghoffer, A.A.; Cerro, R.L.; Abraham, M.A. Catalytic wet oxidation of acetic acid using platinum on alumina monolith catalyst. Catal. Today 1998, 40, 59–71. [Google Scholar] [CrossRef]
- Barbierjr, J.; Delanoë, F.; Jabouille, F.; Duprez, D.; Blanchard, G.; Isnard, P. Total oxidation of acetic acid in aqueous solutions over noble metal catalysts. J. Catal. 1998, 177, 378–385. [Google Scholar] [CrossRef]
- Keav, S.; Martin, A.; Barbier, J.; Duprez, D. Deactivation and reactivation of noble metal catalysts tested in the Catalytic Wet Air Oxidation of phenol. Catal. Today 2010, 151, 143–147. [Google Scholar] [CrossRef]
- Sarno, M.; Sannino, D.; Leone, C.; Ciambelli, P. Evaluating the effects of operating conditions on the quantity, quality and catalyzed growth mechanisms of CNTs. J. Mol. Catal. A Chem. 2012, 357, 26–38. [Google Scholar] [CrossRef]
- Sarno, M.; Cirillo, C.; Ciambelli, P. Selective graphene covering of monodispersed magnetic nanoparticles. Chem. Eng. J. 2014, 246, 27–38. [Google Scholar] [CrossRef]
- Zhao, B.; Shang, C.; Qi, N.; Chen, Z.; Chen, Z. Stability of defects in monolayer MoS2 and their interaction with O2 molecule: A first-principles study. Appl. Surf. Sci. 2017, 412, 385–393. [Google Scholar] [CrossRef]
- Ghafari, M.; Cui, Y.; Alali, A.; Atkinson, J.D. Phenol adsorption and desorption with physically and chemically tailored porous polymers: Mechanistic variability associated with hyper-cross-linking and amination. J. Hazard. Mater. 2019, 361, 162–168. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Value |
|---|---|
| Temperature (°C) | 95 |
| Pressure (MPa) | 0.3 |
| Air Gas Flow (NL/h) | 4.6 |
| Phenol concentration (mg/L) | 4000–1000 |
| Catalyst concentration (g/L) | 0.4 |
| GC-MS Configuration | |
|---|---|
| Injector | |
| Inlet temperature | 230 °C |
| Sample size | 1 µL |
| Split ratio | 12 |
| Column temperature program | |
| Initial temperature | 50 °C for 1 min |
| Rate 1 | 7.0 °C/min to 180 °C for 1 min |
| Rate 2 | 10 °C/min to 300 °C for 1 min |
| Detector | |
| Type | Mass spectrometer |
| Interface temperature | 230 °C |
| Column | |
| Type | HP-5 (0.25 µm × 0.25 mm × 30 m) |
| Flow rate | 1 mL/min |
| Other | |
| Gas Flow | Helium |
| Electron ionization (EI) | 70 eV |
| Scan range | 35 ÷ 450 amu |
| Scan rate | 1.80 scans/s |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaldo, R.; Iuliano, M.; Cocca, M.; Ambrogi, V.; Gentile, G.; Sarno, M. A New Route for Low Pressure and Temperature CWAO: A PtRu/MoS2_Hyper-Crosslinked Nanocomposite. Nanomaterials 2019, 9, 1477. https://doi.org/10.3390/nano9101477
Castaldo R, Iuliano M, Cocca M, Ambrogi V, Gentile G, Sarno M. A New Route for Low Pressure and Temperature CWAO: A PtRu/MoS2_Hyper-Crosslinked Nanocomposite. Nanomaterials. 2019; 9(10):1477. https://doi.org/10.3390/nano9101477
Chicago/Turabian StyleCastaldo, Rachele, Mariagrazia Iuliano, Mariacristina Cocca, Veronica Ambrogi, Gennaro Gentile, and Maria Sarno. 2019. "A New Route for Low Pressure and Temperature CWAO: A PtRu/MoS2_Hyper-Crosslinked Nanocomposite" Nanomaterials 9, no. 10: 1477. https://doi.org/10.3390/nano9101477