Mesoporous Silica-gold Films for Straightforward, Highly Reproducible Monitoring of Mercury Traces in Water

 ,
,  and
and
Abstract
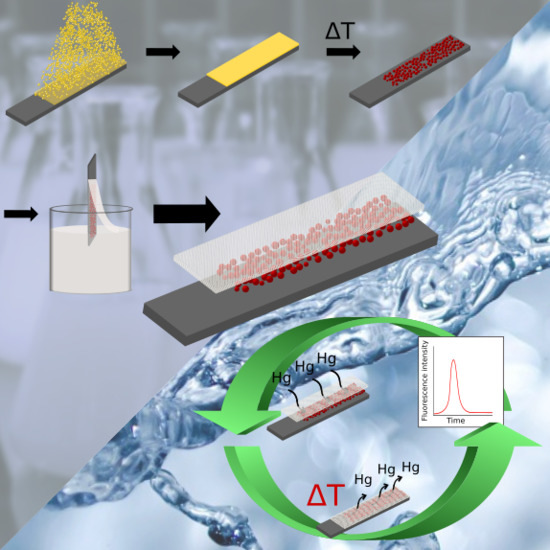
:
1. Introduction
2. Materials and Methods
2.1. Chemicals Used
2.2. Preparation of Gold nanoparticle-covered Silicon Wafers
2.3. Preparation of the Mesoporous Silica Top-layer
2.4. Film Characterization
2.5. Film Stability Investigations and Quantification of Gold Load
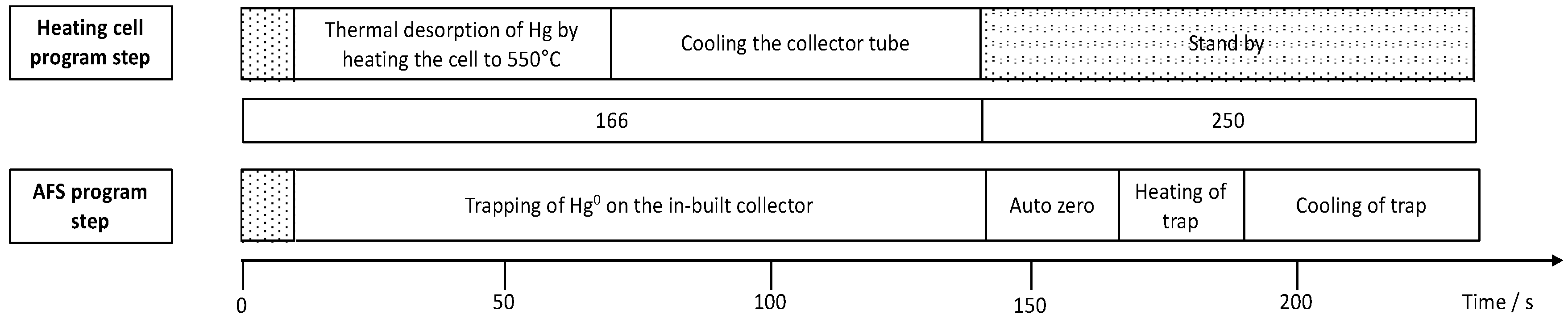
2.6. Mercury Accumulation and Investigation of Analytical Performance for Hg Quantification
3. Results and Discussion
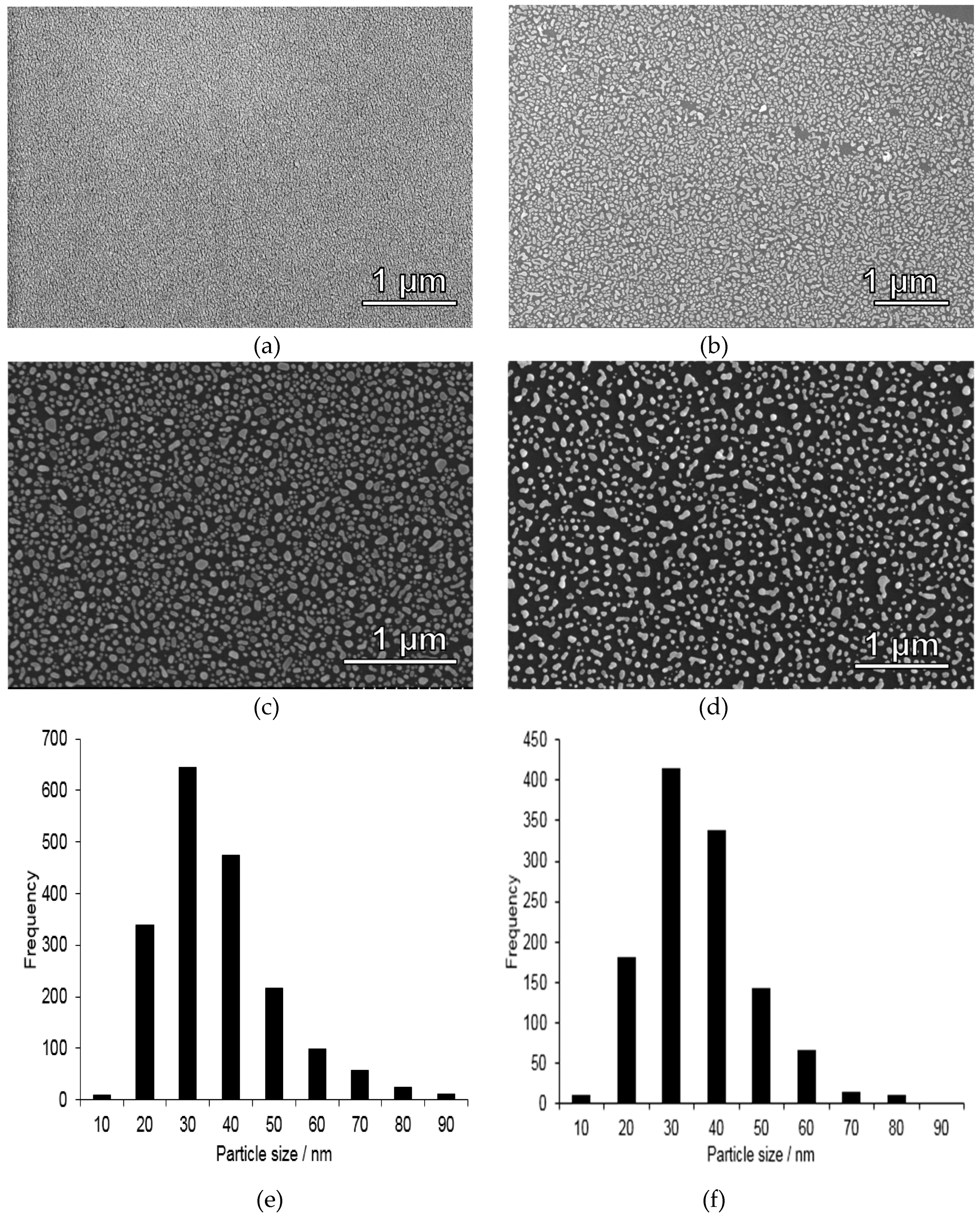
3.1. Synthesis and Characterization of Gold Nanoparticle-coated Substrates
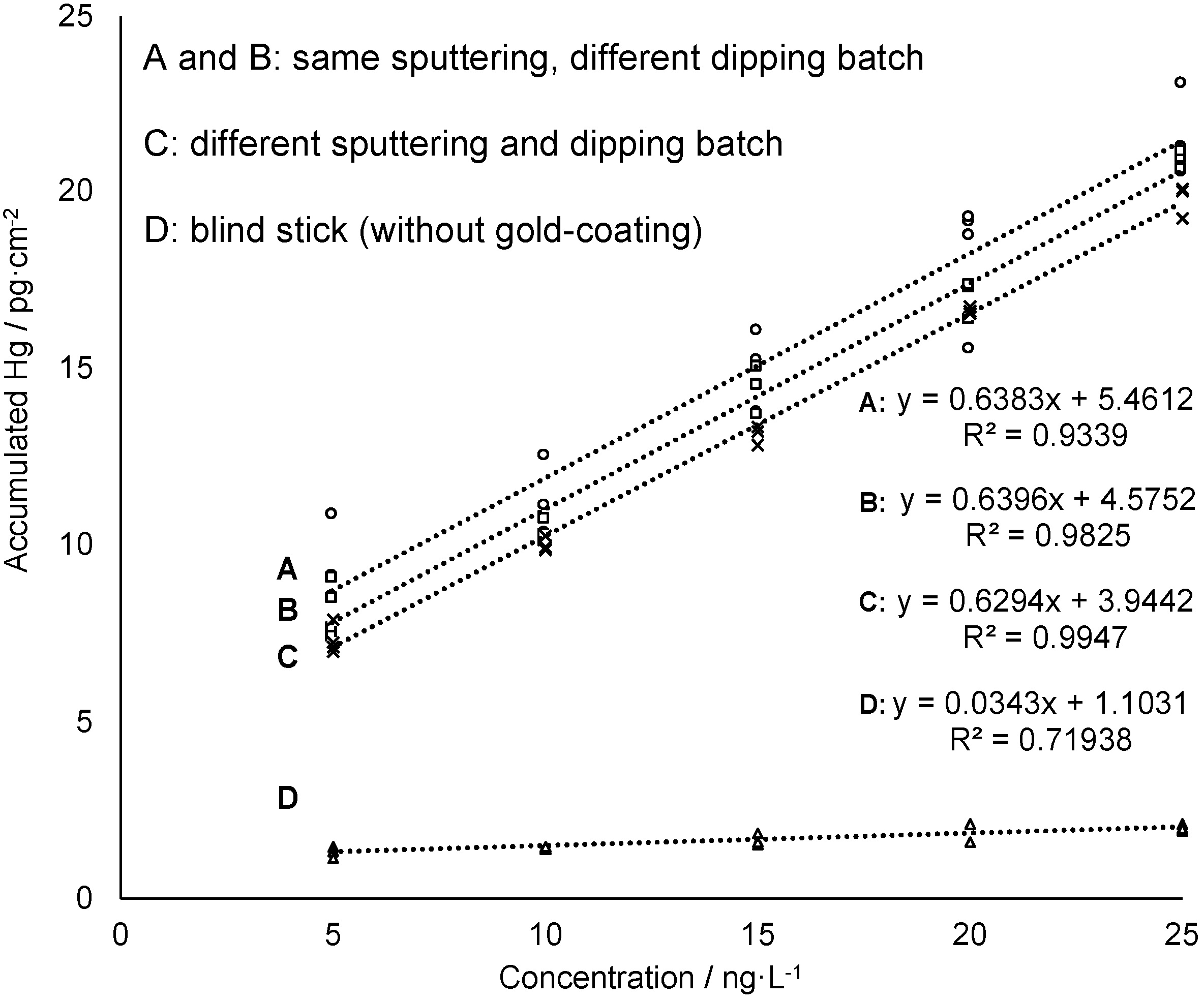
3.2. Mercury Accumulation on the Gold-Mesoporous Silica Films
3.3. Analytical Performance and Validation of Hg Trace Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNEP Minamata Convention on Mercury. Available online: http://www.mercuryconvention.org (accessed on 24 November 2018).
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases and Environmental Transport; UNEP: Geneva, Switzerland, 2013. [Google Scholar]
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, R.B.; Friedli, H.R.; Leaner, J.; Mason, R.; Mukherjee, A.B.; Stracher, G.B.; Streets, D.G.; et al. Global mercury emissions to the atmosphere from anthropogenic and natural sources. Atmos. Chem. Phys. 2010, 10, 5951–5964. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, W.; Zhang, Y.; Tong, Y.; Liu, M.; Wang, H.; Xie, H.; Wang, X. Historical and future trends in global source-receptor relationships of mercury. Sci. Total Environ. 2018, 610–611, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.; Stern, A.H.; Gochfeld, M. Mercury in Commercial Fish: Optimizing Individual Choices to Reduce Risk. Environ. Health Perspect. 2005, 113, 266–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadaro, J.V.; Rabl, A. Global Health Impacts and Costs Due to Mercury Emissions. Risk Anal. 2008, 28, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Directive 2000/60/EC of the European Parliament and of the Council; European Environment Agency: Copenhagen, Denmark, 2000.
- Gill, G.A.; Fitzgerald, W.F. Picomolar mercury measurements in seawater and other materials using stannous chloride reduction and two-stage gold amalgamation with gas phase detection. Mar. Chem. 1987, 20, 227–243. [Google Scholar] [CrossRef]
- Cossa, D.; Sanjuan, J. Automated technique for mercury determination at sub-nanogram per litre levels in natural waters. J. Anal. At. Spectrom. 1995, 10, 287–291. [Google Scholar] [CrossRef]
- Haraldsson, C.; Westerlund, S.; Öhman, P. Determination of mercury in natural samples at the sub-nanogram level using inductively coupled plasma/mass spectrometry after reduction to elemental mercury. Anal. Chim. Acta 1989, 221, 77–84. [Google Scholar] [CrossRef]
- Labatzke, T.; Schlemmer, G. Ultratrace determination of mercury in water following EN and EPA standards using atomic fluorescence spectrometry. Anal. Bioanal. Chem. 2004, 378, 1075–1082. [Google Scholar] [CrossRef]
- Leopold, K.; Foulkes, M.; Worsfold, P. Methods for the determination and speciation of mercury in natural waters—A review. Anal. Chim. Acta 2010, 663, 127–138. [Google Scholar] [CrossRef]
- Li, M.; Gou, H.; Al-Ogaidi, I.; Wu, N. Nanostructured Sensors for Detection of Heavy Metals: A Review. ACS Sustain. Chem. Eng. 2013, 1, 713–723. [Google Scholar] [CrossRef]
- Huber, J.; Leopold, K. Nanomaterial-based strategies for enhanced mercury trace analysis in environmental and drinking waters. TrAC Trends Anal. Chem. 2016, 80, 280–292. [Google Scholar] [CrossRef]
- Panichev, N.; Kalumba, M.M.; Mandiwana, K.L. Solid phase extraction of trace amount of mercury from natural waters on silver and gold nanoparticles. Anal. Chim. Acta 2014, 813, 56–62. [Google Scholar] [CrossRef]
- Parodi, B.; Londonio, A.; Polla, G.; Savio, M.; Smichowski, P. On-line flow injection solid phase extraction using oxidised carbon nanotubes as the substrate for cold vapour-atomic absorption determination of Hg(ii) in different kinds of water. J. Anal. At. Spectrom. 2014, 29, 880–885. [Google Scholar] [CrossRef]
- Yordanova, T.; Dakova, I.; Balashev, K.; Karadjova, I. Polymeric ion-imprinted nanoparticles for mercury speciation in surface waters. Microchem. J. 2014, 113, 42–47. [Google Scholar] [CrossRef]
- Ma, L.; Liu, K.; Yin, M.; Chang, J.; Geng, Y.; Pan, K. Fluorescent nanofibrous membrane (FNFM) for the detection of mercuric ion (II) with high sensitivity and selectivity. Sens. Actuators B Chem. 2017, 238, 120–127. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, J.; Hu, Y.; Zhao, H.; Zhou, J.; Liu, Y.; Lou, Z.; Xu, X. Functional nanomaterials: Study on aqueous Hg(II) adsorption by magnetic Fe3O4@SiO2-SH nanoparticles. J. Taiwan Inst. Chem. Eng. 2016, 60, 394–402. [Google Scholar] [CrossRef]
- Rofouei, M.K.; Rezaei, A.; Masteri-Farahani, M.; Khani, H. Selective extraction and preconcentration of ultra-trace level of mercury ions in water and fish samples using Fe3O4-magnetite-nanoparticles functionalized by triazene compound prior to its determination by inductively coupled plasma-optical emission spectrometry. Anal. Methods 2012, 4, 959–966. [Google Scholar]
- Zierhut, A.; Leopold, K.; Harwardt, L.; Schuster, M. Analysis of total dissolved mercury in waters after on-line preconcentration on an active gold column. Talanta 2010, 81, 1529–1535. [Google Scholar] [CrossRef]
- Zierhut, A.; Leopold, K.; Harwardt, L.; Worsfold, P.; Schuster, M. Activated gold surfaces for the direct preconcentration of mercury species from natural waters. J. Anal. At. Spectrom. 2009, 24, 767–774. [Google Scholar] [CrossRef]
- Faustini, M.; Louis, B.; Albouy, P.A.; Kuemmel, M.; Grosso, D. Preparation of Sol−Gel Films by Dip-Coating in Extreme Conditions. J. Phys. Chem. C 2010, 114, 7637–7645. [Google Scholar] [CrossRef]
- Cagnol, F.; Grosso, D.; Soler-Illia, G.J.; de A.A. Soler-Illia, G.J.; Crepaldi, E.L.; Babonneau, F.; Amenitsch, H.; Sanchez, C. Humidity-controlled mesostructuration in CTAB-templated silica thin film processing. The existence of a modulable steady state. J. Mater. Chem. 2003, 13, 61–66. [Google Scholar] [CrossRef]
- Thompson, C.V. Solid-State Dewetting of Thin Films. Annu. Rev. Mater. Res. 2012, 42, 399–434. [Google Scholar] [CrossRef]
- Bricchi, B.R.; Ghidelli, M.; Mascaretti, L.; Zapelli, A.; Russo, V.; Casari, C.S.; Terraneo, G.; Alessandri, I.; Ducati, C.; Li Bassi, A. Integration of plasmonic Au nanoparticles in TiO2 hierarchical structures in a single-step pulsed laser co-deposition. Mater. Des. 2018, 156, 311–319. [Google Scholar] [CrossRef]
- Tesler, A.B.; Chuntonov, L.; Karakouz, T.; Bendikov, T.A.; Haran, G.; Vaskevich, A.; Rubinstein, I. Tunable Localized Plasmon Transducers Prepared by Thermal Dewetting of Percolated Evaporated Gold Films. J. Phys. Chem. C 2011, 115, 24642–24652. [Google Scholar] [CrossRef]
- Müller, C.M.; Mornaghini, F.C.F.; Spolenak, R. Ordered arrays of faceted gold nanoparticles obtained by dewetting and nanosphere lithography. Nanotechnology 2008, 19, 485306. [Google Scholar] [CrossRef]
- Hubaux, A.; Vos, G. Decision and detection limits for calibration curves. Anal. Chem. 1970, 42, 849–855. [Google Scholar] [CrossRef]
- Horwitz, W. Today’s chemical realities. J. AOAC Int. 1983, 66, 1295–1301. [Google Scholar]
- Thompson, M. The amazing Horwitz function. AMC Tech. Brief 2004, 17, 2p. [Google Scholar]
- Linsinger, T. Comparison of Measurement Result with the Certified Value, Application Note 1; European Commission Joint Research Centre Institute for Reference Materials and Measurements: Brussels, Belgium, 2010. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Found Values |
|---|---|
| Linear working range 1 | 1–100 ng Hg L−1 |
| Regression coefficient R2 | 0.9816 |
| Precision given as RSD | |
| c(Hg) = 1 ng Hg L−1 | 2.39% |
| c(Hg) = 5 ng Hg L−1 | 2.75% |
| c(Hg) = 10 ng Hg L−1 | 2.20% |
| c(Hg) = 25 ng Hg L−1 | 2.45% |
| Accuracy given as recovery in CRM | 92 ± 11% |
| ORMS-5: c(Hg)cert. = 26.2 ± 1.3 ng Hg L−1 | |
| Sampling stick A 2 | 24.1 ± 2.6 ng Hg L−1 |
| Sampling stick B 2 | 24.1 ± 2.5 ng Hg L−1 |
| Sensitivity | |
| Limit of detection 3 | 0.753 ng Hg L−1 |
| Limit of quantification 3 | 1.51 ng Hg L−1 |
| Recyclability of sampling stick | ≥30 cycles |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutschler, A.; Stock, V.; Ebert, L.; Björk, E.M.; Leopold, K.; Lindén, M. Mesoporous Silica-gold Films for Straightforward, Highly Reproducible Monitoring of Mercury Traces in Water. Nanomaterials 2019, 9, 35. https://doi.org/10.3390/nano9010035
Mutschler A, Stock V, Ebert L, Björk EM, Leopold K, Lindén M. Mesoporous Silica-gold Films for Straightforward, Highly Reproducible Monitoring of Mercury Traces in Water. Nanomaterials. 2019; 9(1):35. https://doi.org/10.3390/nano9010035
Chicago/Turabian StyleMutschler, Anna, Vivian Stock, Lena Ebert, Emma M. Björk, Kerstin Leopold, and Mika Lindén. 2019. "Mesoporous Silica-gold Films for Straightforward, Highly Reproducible Monitoring of Mercury Traces in Water" Nanomaterials 9, no. 1: 35. https://doi.org/10.3390/nano9010035