Effect of Microwave Radiation Power on the Size of Aggregates of ZnO NPs Prepared Using Microwave Solvothermal Synthesis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Substrates
2.2. Synthesis of ZnO NPs
2.3. Water Content Analysis
2.4. Morphology Characteristics
2.5. X-ray Powder Diffraction
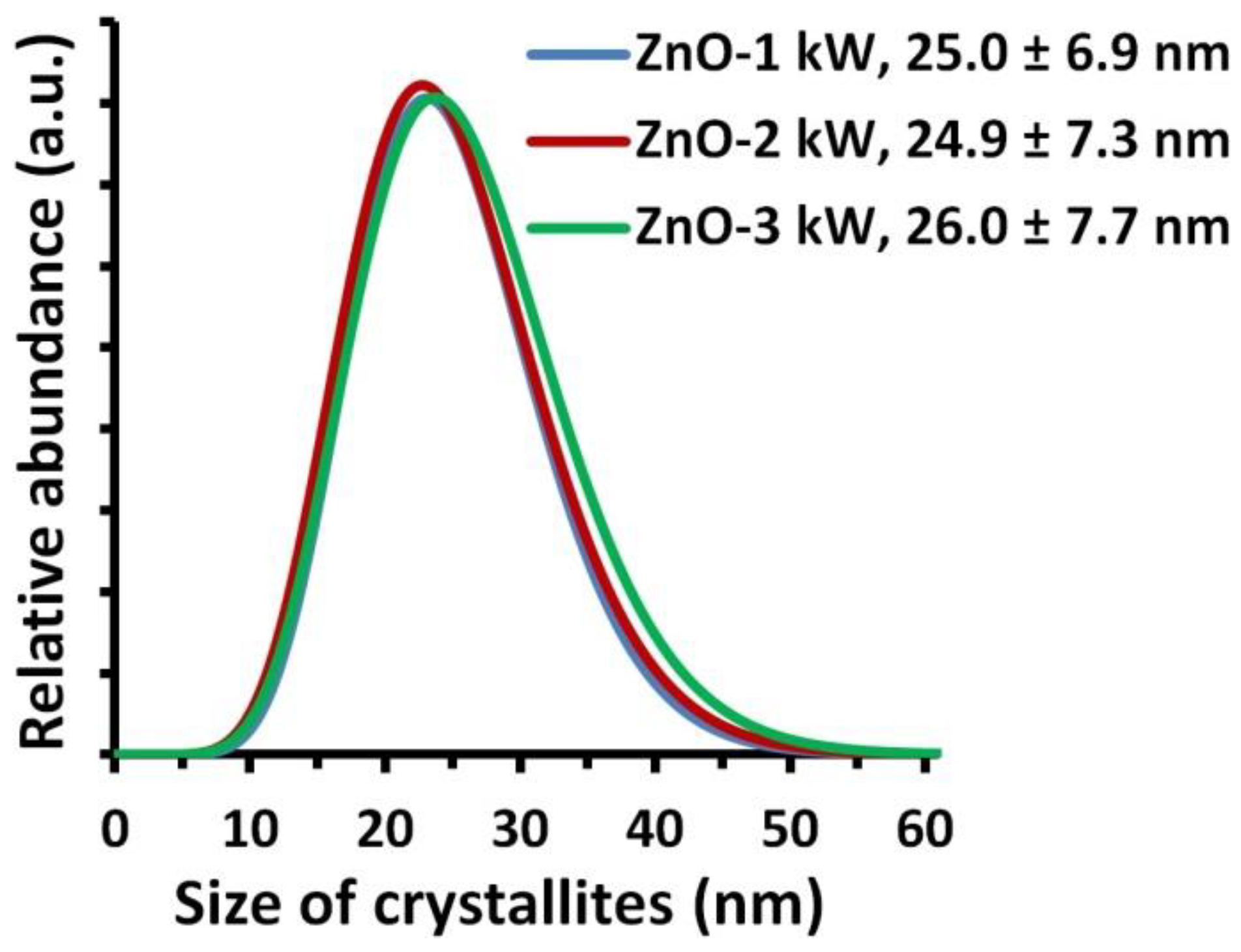
2.6. Crystallite Size Distribution
2.7. Measurement of Density and Specific Surface Area
2.8. Measurement of Average ZnO NPs Size in Water Suspension
3. Results and Discussion
3.1. Morphology
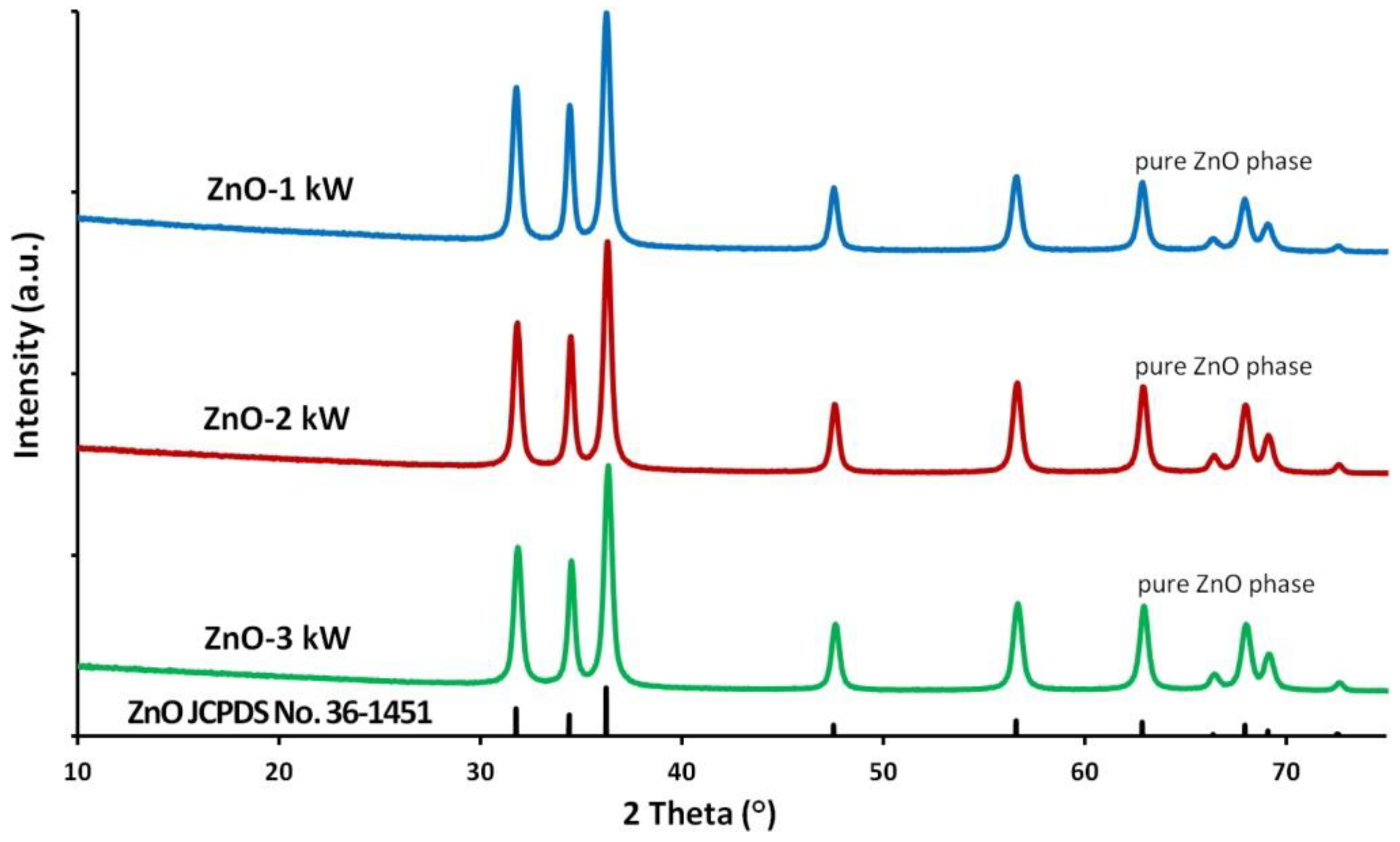
3.2. Phase Composition
3.3. Density, Specific Surface Area and Average Size and Size Distribution of NPs
- ZnO NPs are built of single crystals.
- ZnO NPs size is equal to the crystallite size.
- The change of microwave radiation power did not impact the average size of the obtained ZnO NPs.
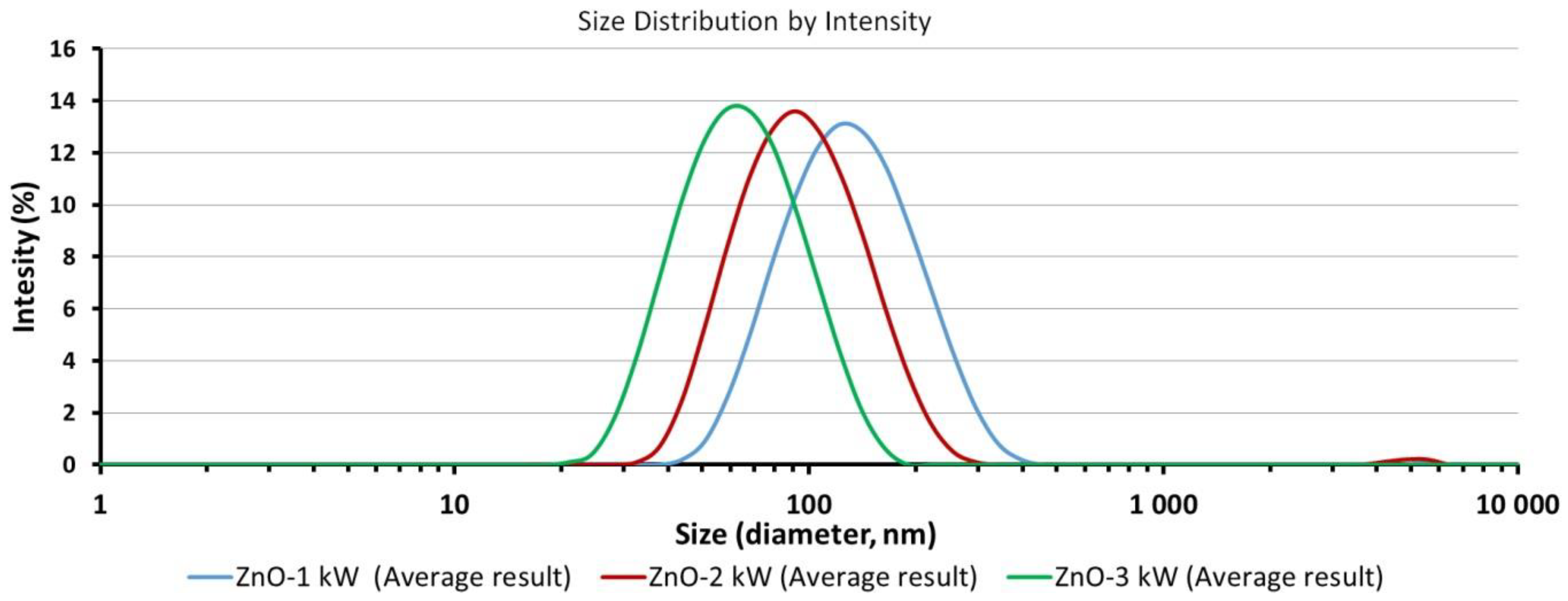
3.4. Average Size and Size Distribution of ZnO NPs in Water Suspensions
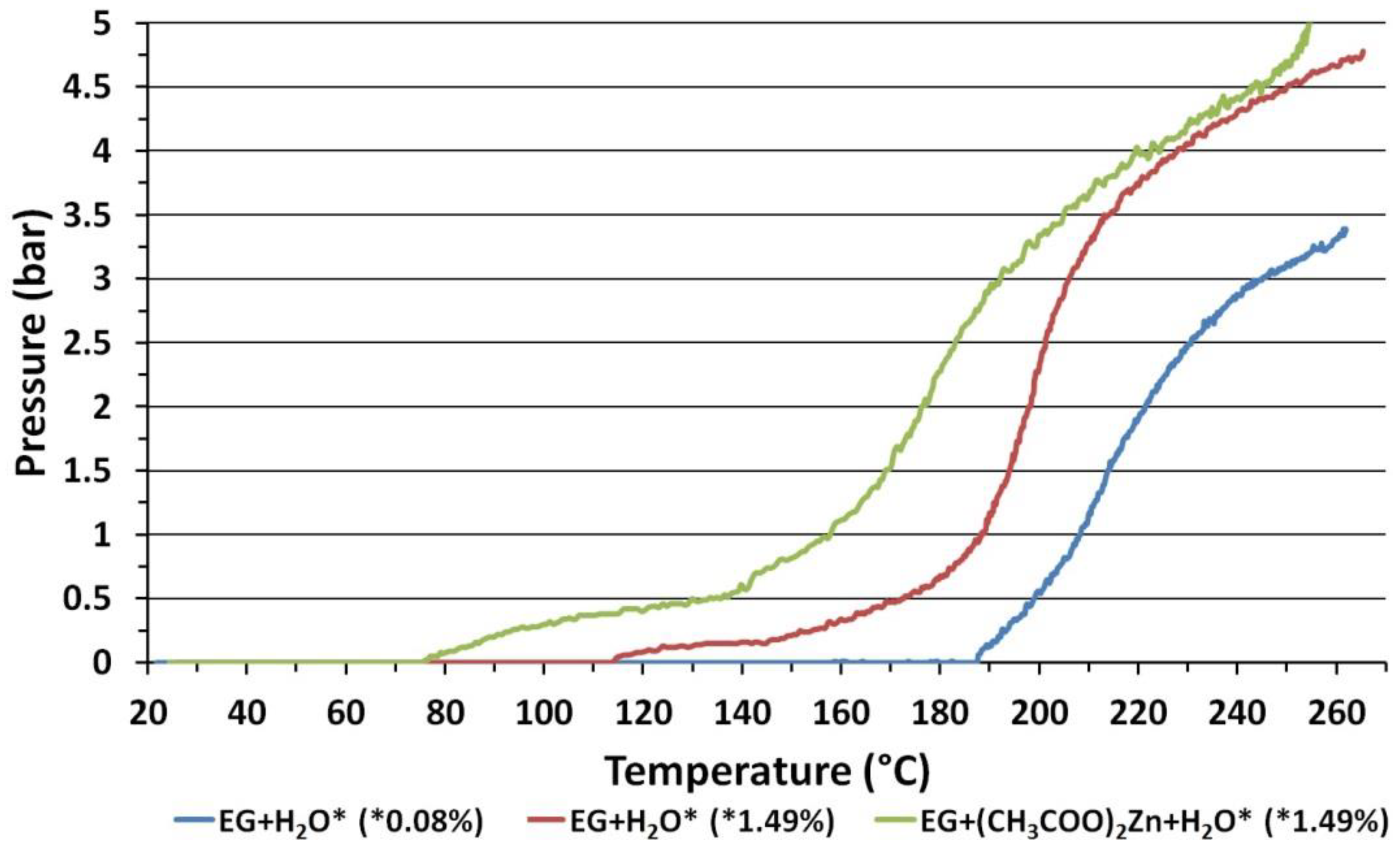
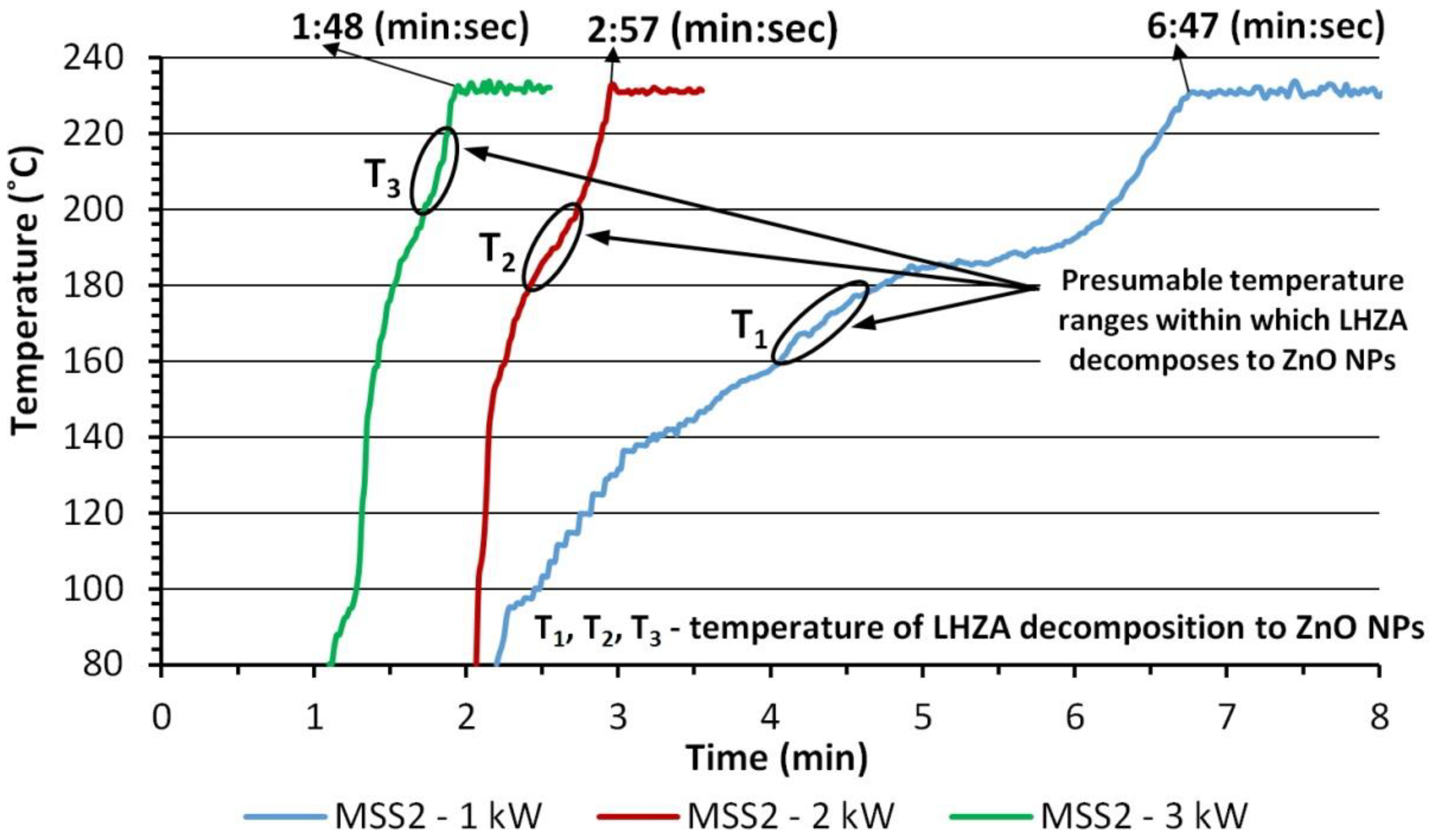
3.5. Dependence of Pressure on Ethylene Glycol Temperature
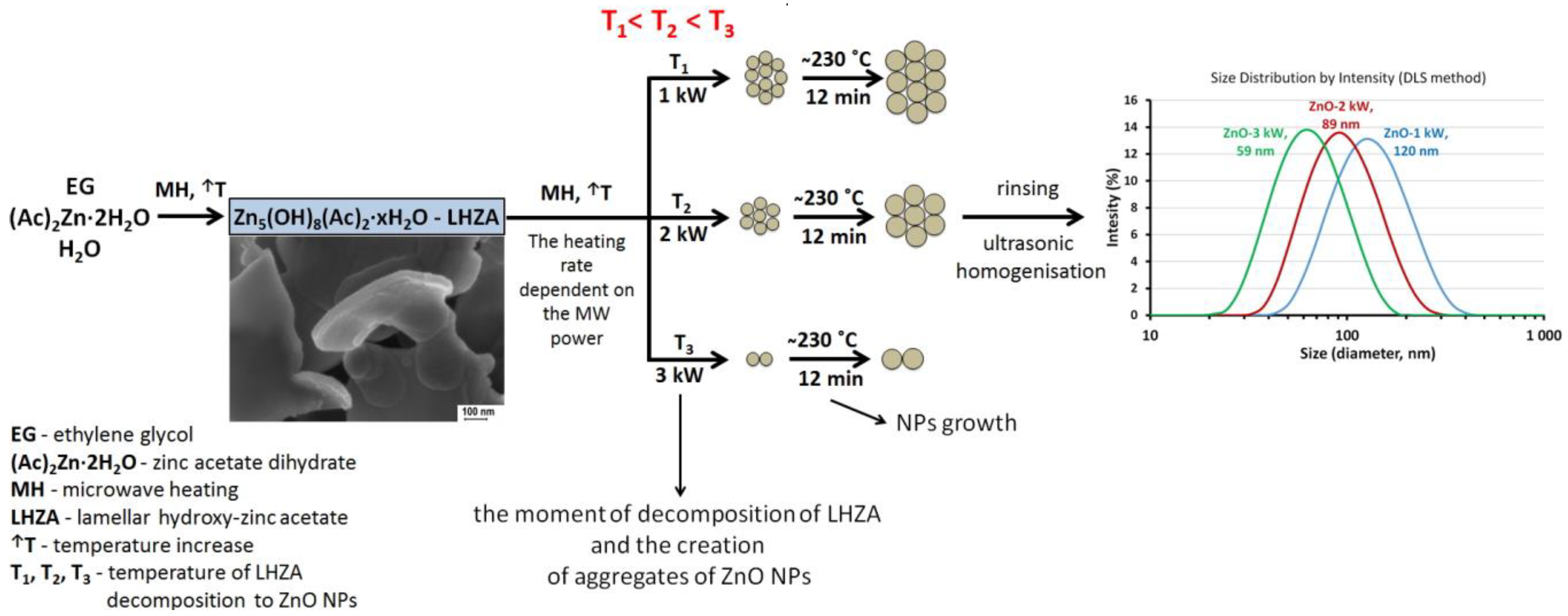
3.6. ZnO NPs Synthesis Temperature
- First stage is the duration of feedstock pre-heating until synthesis parameters are reached;
- Second stage is the heating duration for constant synthesis parameters;
- Third stage is the duration of reactor cooling down.
- The size of ZnO NPs in the MSS is the function of H2O content in the zinc acetate solution in ethylene glycol.
- The size of ZnO NPs increases until the unreacted zinc acetate is exhausted.
- The solvent used by us, ethylene glycol, acts as a stabilizing agent, which eliminates uncontrolled particle growth.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klingshirn, C.F.; Waag, A.; Hoffmann, A.; Geurts, J. Zinc Oxide, 1st ed.; Springer: Berlin, Germany, 2010; ISBN 978-3-642-10576-0. [Google Scholar]
- Ozgur, U.; Hofstetter, D.; Morkoc, H. ZnO devices and applications: A review of current status and future prospects. Proc. IEEE 2010, 98, 1255–1268. [Google Scholar] [CrossRef]
- Zhang, Y.; Nayak, T.R.; Hong, H.; Cai, W. Biomedical Applications of Zinc Oxide Nanomaterials. Curr. Mol. Med. 2013, 13, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carmona, M.; Gun’ko, Y.; Vallet-Regí, M. ZnO Nanostructures for Drug Delivery and Theranostic Applications. Nanomaterials 2018, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Pokrowiecki, R.; Pałka, K.; Mielczarek, A. Nanomaterials in dentistry: A cornerstone or a black box? Nanomedicine 2018, 13, 639–667. [Google Scholar] [CrossRef] [PubMed]
- Cierech, M.; Osica, I.; Kolenda, A.; Wojnarowicz, J.; Szmigiel, D.; Łojkowski, W.; Kurzydłowski, K.; Ariga, K.; Mierzwińska-Nastalska, E. Mechanical and Physicochemical Properties of Newly Formed ZnO-PMMA Nanocomposites for Denture Bases. Nanomaterials 2018, 8, 305. [Google Scholar] [CrossRef] [PubMed]
- Cierech, M.; Kolenda, A.; Grudniak, A.M.; Wojnarowicz, J.; Woźniak, B.; Gołaś, M.; Swoboda-Kopeć, E.; Łojkowski, W.; Mierzwińska-Nastalska, E. Significance of polymethylmethacrylate (PMMA) modification by zinc oxide nanoparticles for fungal biofilm formation. Int. J. Pharm. 2016, 510, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Osmond, M.J.; McCall, M.J. Zinc oxide nanoparticles in modern sunscreens: An analysis of potential exposure and hazard. Nanotoxicology 2016, 4, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Paszek, E.; Czyz, J.; Woźniacka, O.; Jakubiak, D.; Wojnarowicz, J.; Łojkowski, W.; Stępień, E. Zinc oxide nanoparticles impair the integrity of human umbilical vein endothelial cell monolayer in vitro. J. Biomed. Nanotechnol. 2012, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Coleman, V.A.; Jagadish, C. Basic Properties and Applications of ZnO. In Zinc Oxide Bulk, Thin Films and Nanostructures, 1st ed.; Jagadish, C., Pearton, S., Eds.; Elsevier: Oxford, UK, 2006; pp. 1–20. ISBN 978-0-08-044722-3. [Google Scholar]
- Kołodziejczak-Radzimska, A.; Jesionowski, T. Zinc Oxide-From Synthesis to Application: A Review. Materials 2014, 7, 2833–2881. [Google Scholar] [CrossRef] [PubMed]
- Cierech, M.; Wojnarowicz, J.; Szmigiel, D.; Bączkowski, B.; Grudniak, A.; Wolska, K.; Łojkowski, W.; Mierzwińska-Nastalska, E. Preparation and characterization of ZnO-PMMA resin nanocomposites for denture bases. Acta Bioeng. Biomech. 2016, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Salzano de Luna, M.; Galizia, M.; Wojnarowicz, J.; Rosa, R.; Lojkowski, W.; Acierno, D.; Filippone, G.; Leonelli, C. Dispersing hydrophilic nanoparticles in hydrophobic polymers: HDPE/ZnO nanocomposites by a novel template-based approach. Express Polym. Lett. 2014, 8, 362–372. [Google Scholar] [CrossRef] [Green Version]
- The Global Market for Zinc Oxide Nanoparticles; ID: 3833830; Future Markets, Inc.: Edinburgh, UK, 2016.
- The Global Market for Nanotechnology and Nanomaterials in Cosmetics; Personal Care and Sunscreens, ID: 3784908; Future Markets, Inc.: Edinburgh, UK, 2016.
- Omelchenko, M.M.; Wojnarowicz, J.; Salamonczyk, M.; Lojkowski, W. Lyotropic liquid crystal based on zinc oxide nanoparticles obtained by microwave solvothermal synthesis. Mater. Chem. Phys. 2017, 192, 383–391. [Google Scholar] [CrossRef]
- Sabir, S.; Arshad, M.; Chaudhari, S.K. Zinc Oxide Nanoparticles for Revolutionizing Agriculture: Synthesis and Applications. Sci. World J. 2014, 2014, 925494. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, J.; Opalinska, A.; Chudoba, T.; Gierlotka, S.; Mukhovskyi, R.; Pietrzykowska, E.; Sobczak, K.; Lojkowski, W. Effect of water content in ethylene glycol solvent on the size of ZnO nanoparticles prepared using microwave solvothermal synthesis. J. Nanomater. 2016, 2016, 2789871. [Google Scholar] [CrossRef]
- Wojnarowicz, J.; Chudoba, T.; Koltsov, I.; Gierlotka, S.; Dworakowska, S.; Lojkowski, W. Size control mechanism of ZnO nanoparticles obtained in microwave solvothermal synthesis. Nanotechnology 2018, 29, 065601. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, J.; Chudoba, T.; Gierlotka, S.; Sobczak, K.; Lojkowski, W. Size Control of Cobalt-Doped ZnO Nanoparticles Obtained in Microwave Solvothermal Synthesis. Crystals 2018, 8, 179. [Google Scholar] [CrossRef]
- Lojkowski, W.; Leonelli, C.; Chudoba, T.; Wojnarowicz, J.; Majcher, A.; Mazurkiewicz, A. High-Energy-Low-Temperature Technologies for the Synthesis of Nanoparticles: Microwaves and High Pressure. Inorganics 2014, 2, 606–619. [Google Scholar] [CrossRef]
- Kusnieruk, S.; Wojnarowicz, S.; Chodara, A.; Chudoba, T.; Gierlotka, S.; Lojkowski, W. Influence of hydrothermal synthesis parameters on the properties of hydroxyapatite nanoparticles. Beilstein J. Nanotechnol. 2016, 7, 1586–1601. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, J.; Chudoba, T.; Majcher, A.; Łojkowski, W. Microwaves Applied to Hydrothermal Synthesis of Nanoparticles. In Microwave Chemistry, 1st ed.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2017; pp. 205–224. ISBN 9783110479935. [Google Scholar]
- Pimentel, A.; Samouco, A.; Nunes, D.; Araújo, A.; Martins, R.; Fortunato, E. Ultra-Fast Microwave Synthesis of ZnO Nanorods on Cellulose Substrates for UV Sensor Applications. Materials 2017, 10, 1308. [Google Scholar] [CrossRef] [PubMed]
- Schanche, J.S. Microwave synthesis solutions from Personal Chemistry. Mol. Divers. 2003, 7, 291–298. [Google Scholar] [CrossRef]
- Rizzuti, A.; Leonelli, C. Crystallization of aragonite particles from solution under microwave irradiation. Powder Technol. 2008, 186, 255–262. [Google Scholar] [CrossRef]
- Hasanpoor, M.; Aliofkhazraei, M.; Delavari, H. Microwave-assisted Synthesis of Zinc Oxide Nanoparticles. Proc. Mater. Sci. 2015, 11, 320–325. [Google Scholar] [CrossRef]
- Pimentel, A.; Ferreira, S.H.; Nunes, D.; Calmeiro, T.; Martins, R.; Fortunato, E. Microwave Synthesized ZnO Nanorod Arrays for UV Sensors: A Seed Layer Annealing Temperature Study. Materials 2016, 9, 299. [Google Scholar] [CrossRef] [PubMed]
- Bilecka, I.; Elser, P.; Niederberger, M. Kinetic and Thermodynamic Aspects in the Microwave-Assisted Synthesis of ZnO Nanoparticles in Benzyl Alcohol. ACS Nano 2009, 3, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.U.; Kang, M.; Kim, H.S. Microwave-assisted Facile and Ultrafast Growth of ZnO Nanostructures and Proposition of Alternative Microwave-assisted Methods to Address Growth Stoppage. Sci. Rep. 2016, 6, 24870. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.P.; Morales, G.; Quintanilla Ma, L.L. Microwave Assisted Synthesis of ZnO Nanoparticles: Effect of Precursor Reagents, Temperature, Irradiation Time, and Additives on Nano-ZnO Morphology Development. J. Mater. 2013, 2013, 478681. [Google Scholar] [CrossRef]
- Pimentel, A.; Nunes, D.; Duarte, P.; Rodrigues, J.; Costa, F.M.; Monteiro, T.; Martins, R.; Fortunato, E. Synthesis of Long ZnO Nanorods under Microwave Irradiation or Conventional Heating. J. Phys. Chem. C 2014, 118, 14629–14639. [Google Scholar] [CrossRef]
- Glaspell, G.; Dutta, P.; Manivannan, A. A Room-Temperature and Microwave Synthesis of M-Doped ZnO (M = Co, Cr, Fe, Mn & Ni). J. Clust. Sci. 2005, 16, 523–536. [Google Scholar] [CrossRef]
- Lojkowski, W.; Gedanken, A.; Grzanka, E.; Opalinska, A.; Strachowski, T.; Pielaszek, R.; Tomaszewska-Grzeda, A.; Yatsunenko, S.; Godlewski, M.; Matysiak, H.; et al. Solvothermal synthesis of nanocrystalline zinc oxide doped with Mn2+, Ni2+, Co2+ and Cr3+ ions. J. Nanopart. Res. 2009, 11, 1991–2002. [Google Scholar] [CrossRef]
- Liang, B.; Feng, Y.; Zhang, Y. ZnO whiskers synthesized by rapid microwave reduction reaction. Int. J. Mater. Res. 2017, 108, 611–614. [Google Scholar] [CrossRef]
- Strachowski, T.; Grzanka, E.; Palosz, B.F.; Presz, A.; Ślusarski, L.; Łojkowski, W. Microwave Driven Hydrothermal Synthesis of Zinc Oxide Nanopowders. Solid State Pheom. 2003, 94, 189–192. [Google Scholar] [CrossRef]
- Pimentel, A.; Rodrigues, J.; Duarte, P.; Nunes, D.; Costa, F.M.; Monteiro, T.; Martins, R.; Fortunato, E. Effect of solvents on ZnO nanostructures synthesized by solvothermal method assisted by microwave radiation: A photocatalytic study. J. Mater. Sci. 2015, 50, 5777–5787. [Google Scholar] [CrossRef]
- Krishnapriya, R.; Praneetha, S.; Vadivel Murugan, A. Energy-efficient, microwave-assisted hydro/solvothermal synthesis of hierarchical flowers and rice grain-like ZnO nanocrystals as photoanodes for high performance dye-sensitized solar cells. CrystEngComm 2015, 17, 8353–8367. [Google Scholar] [CrossRef]
- Chieng, B.W.; Loo, Y.Y. Synthesis of ZnO nanoparticles by modified polyol method. Mater. Lett. 2012, 73, 78–82. [Google Scholar] [CrossRef]
- Lian, J.; Liang, Y.; Kwong, F.-L.; Ding, Z.; Ng, D.H.L. Template-free solvothermal synthesis of ZnO nanoparticles with controllable size and their size-dependent optical. Mater. Lett. 2012, 66, 318–320. [Google Scholar] [CrossRef]
- Wang, H.; Xie, C.; Zeng, D. Controlled growth of ZnO by adding H2O. J. Cryst. Growth 2005, 277, 372–377. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, S.; Kim, D.; Hwang, S.; Jeon, M.; Moona, J. ZnO nanoparticles with controlled shapes and sizes prepared using a simple polyol synthesis. Superlattic. Microst. 2008, 43, 330–339. [Google Scholar] [CrossRef]
- Phimmavong, K.; Song, J.H.; Cho, S.B.; Lim, D.Y. Effect of Water Content on the Morphology of ZnO Powders Synthesized in Binary Solvent Mixtures by Glycol Process. J. Korean Ceram. Soc. 2017, 54, 211–216. [Google Scholar] [CrossRef]
- Nichols, G.; Byard, S.; Bloxham, M.J.; Botterill, J.; Dawson, N.J.; Dennis, A.; Diart, V.; North, N.C.; Sherwood, J.D. A review of the terms agglomerate and aggregate with a recommendation for nomenclature used in powder and particle characterization. J. Pharm. Sci. 2002, 91, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Faure, B.; Salazar-Alvarez, G.; Ahniyaz, G.; Villaluenga, I.; Berriozabal, G.; De Miguel, Y.R.; Bergström, L. Dispersion and surface functionalization of oxide nanoparticles for transparent photocatalytic and UV-protecting coatings and sunscreens. Sci. Technol. Adv. Mater. 2013, 14, 023001. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Nanotechnologies—Terminology and Definitions for Nano-Objects—Nanoparticle, Nanofibre and Nanoplate; ISO/TS 27687:2008; ISO: Geneva, Switzerland, 2008. [Google Scholar]
- International Organization for Standardization. Nanotechnologies—Vocabulary—Part 2: Nano-Objects; ISO/TS 80004-2:2015; ISO: Geneva, Switzerland, 2015. [Google Scholar]
- International Organization for Standardization. Nanotechnologies—Plain Language Explanation of Selected Terms from the ISO/IEC 80004 Series; ISO/TR 18401:2017; ISO: Geneva, Switzerland, 2017. [Google Scholar]
- Wong, S.W.; Leung, P.T.; Djurisić, A.B.; Leung, K.M. Toxicities of nano zinc oxide to five marine organisms: Influences of aggregate size and ion solubility. Anal. Bioanal. Chem. 2010, 396, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Ribeiro, F.; Wojnarowicz, J.; Łojkowski, W.; Jurkschat, K.; Crossley, A.; Soares, A.M.; Loureiro, S. Zinc oxide nanoparticles toxicity to Daphnia magna: Size-dependent effects and dissolution. Environ. Toxicol. Chem. 2014, 33, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Cheng, W.L.; Huang, S.C.; Chen, Y.P.; Chou, H.K.; Cheng, H.F. Characterizing titanium dioxide and zinc oxide nanoparticles in sunscreen spray. Int. J. Cosmet. Sci. 2015, 37, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Leonard, J.P.; Nettleship, I.; Lee, J.K.; Soong, Y.; Martello, D.V.; Chyu, M.K. Characterization of ZnO nanoparticle suspension in water: Effectiveness of ultrasonic dispersion. Powder Technol. 2009, 194, 75–80. [Google Scholar] [CrossRef]
- Woźniak, B.; Dąbrowska, S.; Wojnarowicz, J.; Chudoba, T.; Łojkowski, W. Content available remote Coating synthetic materials with zinc oxide nanoparticles acting as a UV filter. Glass Ceram. 2017, 3, 15–17. [Google Scholar]
- Guo, H.; He, H.; Hu, C.; Tian, Y.; Xi, Y.; Chen, J.; Tian, L. Effect of Particle Size in Aggregates of ZnO-Aggregate-Based Dye-Sensitized Solar Cells. Electrochim. Acta 2014, 120, 23–29. [Google Scholar] [CrossRef]
- Zhang, Q.; Park, K.; Cao, G. Synthesis of ZnO Aggregates and Their Application in Dye-sensitized Solar Cells. Mater. Matters 2014, 5, 32–39. [Google Scholar]
- Shi, J.; Liu, Y.; Peng, Q. ZnO hierarchical aggregates: Solvothermal synthesis and application in dye-sensitized solar cells. Nano Res. 2013, 6, 441–448. [Google Scholar] [CrossRef]
- Lu, H.H.; Zheng, Y.Z.; Zhao, J.; Chen, J.F.; Tao, X. Aqueous chemical synthesis of large-scale ZnO aggregates with high-efficient light-scattering and application in dye-sensitized solar cells. Electrochim. Acta 2013, 90, 649–655. [Google Scholar] [CrossRef]
- Šarić, A.; Štefanić, G.; Dražić, G.; Gotića, M. Solvothermal synthesis of zinc oxide microspheres. J. Alloys Compd. 2015, 652, 91–99. [Google Scholar] [CrossRef]
- Saito, N.; Matsumoto, K.; Watanabe, K.; Aubert, T.; Grasset, F.; Sakaguchi, I.; Haneda, H. Solvothermal synthesis of ZnO spherical particles and VOC sensor application. J. Ceram. Soc. Jpn. 2014, 122, 488–491. [Google Scholar] [CrossRef]
- Zhang, F. “Wet” Chemical Synthesis and Manipulation of Upconversion Nanoparticles. In Photon Upconversion Nanomaterials, 1st ed.; Springer: Berlin, Germany, 2015; pp. 21–71. [Google Scholar]
- Kamiya, H.; Iijima, M. Surface modification and characterization for dispersion stability of inorganic nanometer-scaled particles in liquid media. Sci. Technol. Adv. Mater. 2010, 11, 044304. [Google Scholar] [CrossRef] [PubMed]
- Mandzy, N.; Grulke, E.; Druffel, T. Breakage of TiO2 agglomerates in electrostatically stabilized aqueous dispersions. Powder Technol. 2012, 160, 121–126. [Google Scholar] [CrossRef]
- Majcher, A.; Wiejak, J.; Przybylski, J.; Chudoba, T.; Wojnarowicz, J. A Novel Reactor for Microwave Hydrothermal Scale-up Nanopowder Synthesis. Int. J. Chem. React. Eng. 2013, 11, 361–368. [Google Scholar] [CrossRef]
- Wojnarowicz, J.; Chudoba, T.; Smoleń, D.; Łojkowski, W.; Majcher, A.; Mazurkiewicz, A. Examples of the Nanoparticles Produced by Microwave Solvothermal Synthesis (MSS) Route. Glass Ceram. 2014, 6, 8–11. [Google Scholar]
- Pielaszek, R. FW15/45M method for determination of the grain size distribution from powder diffraction line profile. J. Alloys Compd. 2004, 37, 128–132. [Google Scholar] [CrossRef]
- Nanopowder XRD Processor Demo. Available online: http://science24.com/xrd/ (accessed on 10 January 2018).
- FW1/5 4/5M Method of Evaluation of Grain Size Distribution by Powder Diffraction. Available online: http://science24.com/fw145m/ (accessed on 10 January 2018).
- Promnopasa, W.; Thongtem, T.; Thongtem, S. Effect of microwave power on energy gap of ZnO nanoparticles synthesized by microwaving through aqueous solutions. Superlattic. Microst. 2015, 78, 71–78. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Synthesis Duration (min) | Synthesis Pressure (bar) | Microwave Power (kW) | Feedstock Volume (mL) | Power Per 1 mL of Feedstock (W/mL) |
|---|---|---|---|---|---|
| ZnO-1 kW | 12 | 4 | 1 | 270 | 3.7 |
| ZnO-2 kW | 12 | 4 | 2 | 270 | 7.4 |
| ZnO-3 kW | 12 | 4 | 3 | 270 | 11.1 |
| Sample Name | Specific Surface Area, as ± σ (m2/g) | Skeleton Density, ρs ± σ (g/cm3) | Average Particle Size from SSA, d (nm) | Average Crystallite Size, Scherrer’s Formula, da, dc (nm) | Average Crystallite Size, Nanopowder XRD Processor Demo, d ± σ (nm) |
|---|---|---|---|---|---|
| ZnO-1 kW | 40.1 ± 0.1 | 5.16 ± 0.06 | 29.0 | 21a; 29c | 25.0 ± 6.9 |
| ZnO-2 kW | 40.6 ± 0.1 | 5.19 ± 0.03 | 28.5 | 24a; 28c | 24.9 ± 7.3 |
| ZnO-3 kW | 40.6 ± 0.1 | 5.18 ± 0.03 | 28.6 | 25a; 30c | 26.0 ± 7.7 |
| Suspension Name | ZnO NPs Concentration in H2O (%) | Average Diameter, (nm) | Polydispersity Index, PI | Distribution Type | Average Size for Peak–Peak Intensity (nm-%) |
|---|---|---|---|---|---|
| ZnO-1 kW | 0.05 | 120 | 0.149 | Bimodal | 141-99.8; 4714-0.2 |
| ZnO-2 kW | 0.05 | 89 | 0.156 | Bimodal | 102-99.4; 4909-0.6 |
| ZnO-3 kW | 0.05 | 59 | 0.141 | Monomodal | 68-100 |
| Sample Name | 1st Stage—Pre-Heating Duration, 0→4 bar, (min:s) | 2nd Stage—Heating Duration, 4 bar, ~230 °C, (min:s) | 3rd Stage—Cooling down Duration, ~230 °C→~90 °C | Total Synthesis Duration (min:s) |
|---|---|---|---|---|
| ZnO-1 kW | 6:47 | 12:00 | a few seconds | ~18:47 |
| ZnO-2 kW | 2:57 | 12:00 | a few seconds | ~14:57 |
| ZnO-3 kW | 1:48 | 12:00 | a few seconds | ~13:48 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojnarowicz, J.; Chudoba, T.; Gierlotka, S.; Lojkowski, W. Effect of Microwave Radiation Power on the Size of Aggregates of ZnO NPs Prepared Using Microwave Solvothermal Synthesis. Nanomaterials 2018, 8, 343. https://doi.org/10.3390/nano8050343
Wojnarowicz J, Chudoba T, Gierlotka S, Lojkowski W. Effect of Microwave Radiation Power on the Size of Aggregates of ZnO NPs Prepared Using Microwave Solvothermal Synthesis. Nanomaterials. 2018; 8(5):343. https://doi.org/10.3390/nano8050343
Chicago/Turabian StyleWojnarowicz, Jacek, Tadeusz Chudoba, Stanisław Gierlotka, and Witold Lojkowski. 2018. "Effect of Microwave Radiation Power on the Size of Aggregates of ZnO NPs Prepared Using Microwave Solvothermal Synthesis" Nanomaterials 8, no. 5: 343. https://doi.org/10.3390/nano8050343