A DFT Screening of M-HKUST-1 MOFs for Nitrogen-Containing Compounds Adsorption

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Bulk Structure
3.2. Molecular Adsorption
3.2.1. NO Adsorption
3.2.2. NH3 Adsorption
3.2.3. NO2 Adsorption
3.3. Dissociative Adsorption
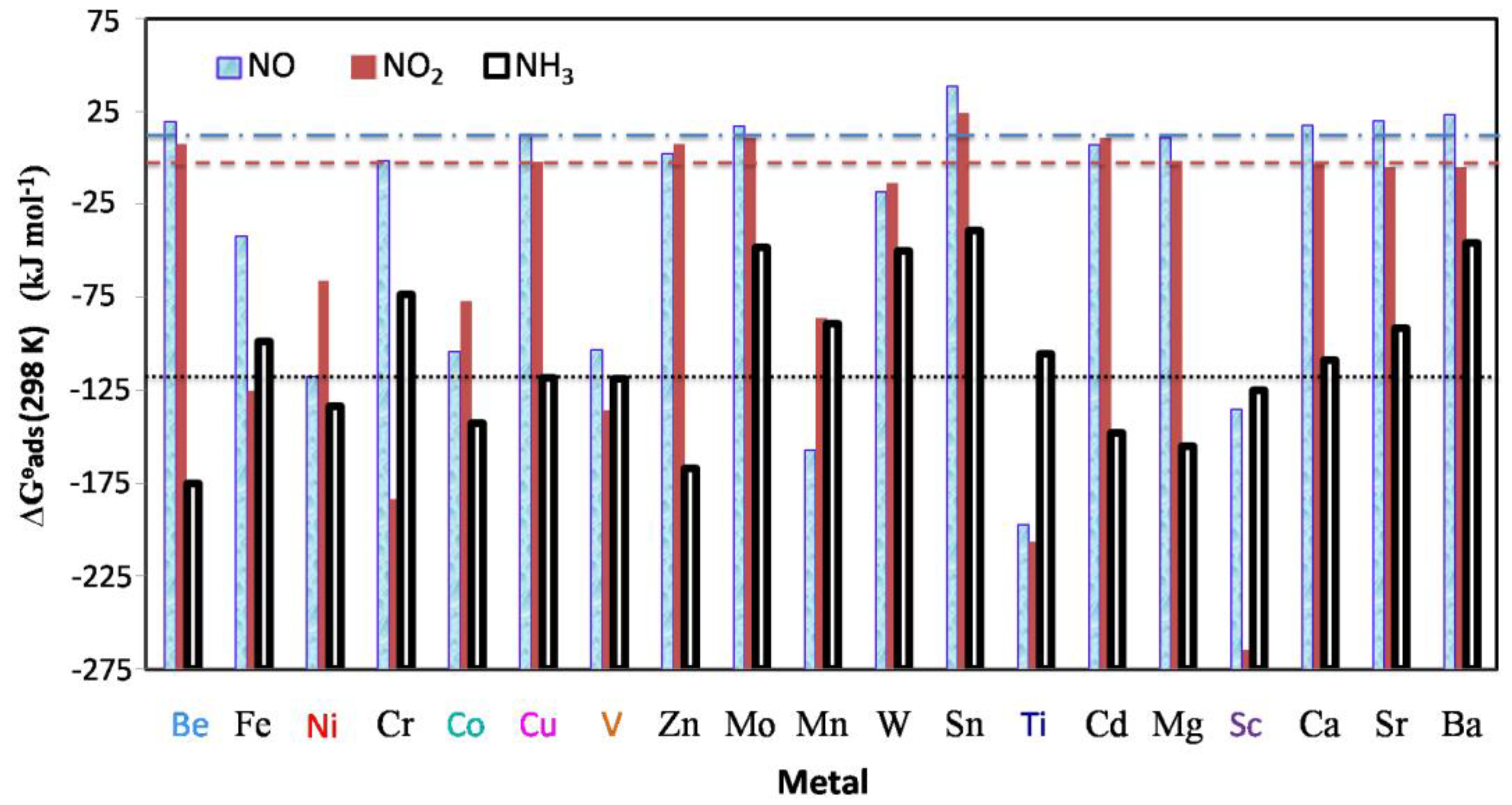
3.4. Thermodynamics
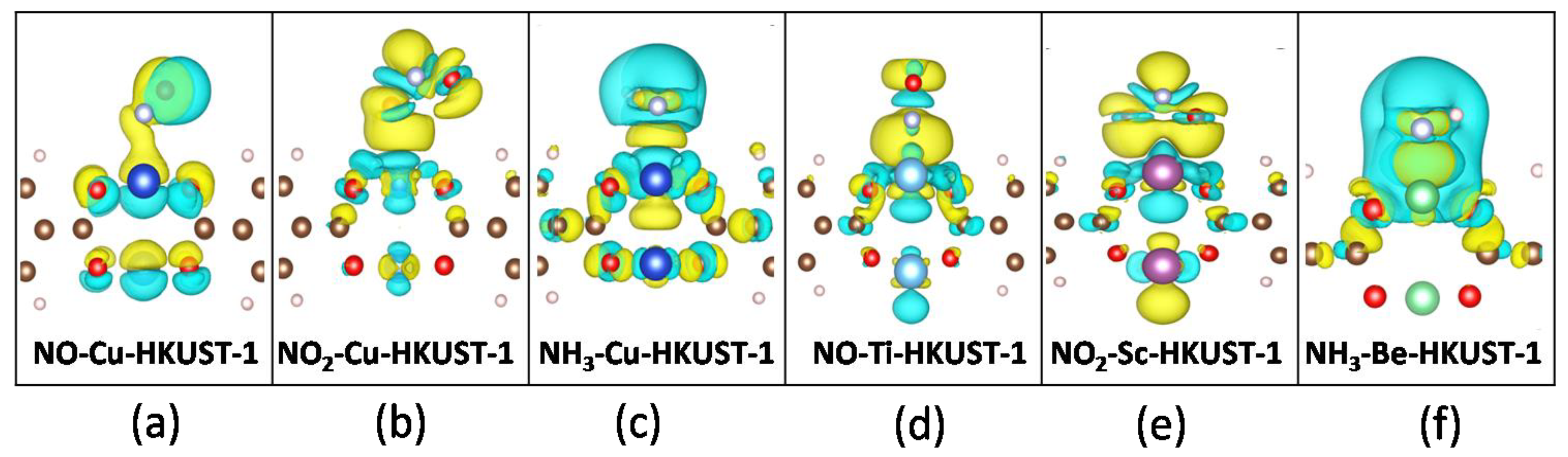
3.5. Analysis of Electronic Properties
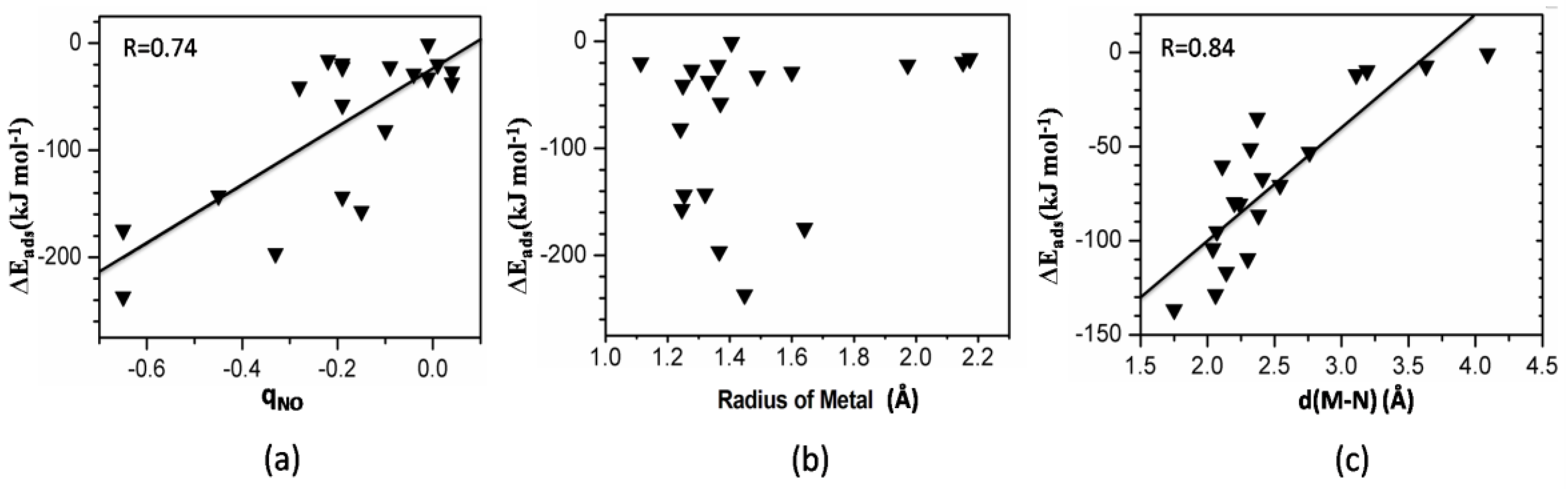
3.6. Correlations
3.6.1. Adsorption Energy and Bader Charge
3.6.2. Adsorption Energy and Metal Radius
3.6.3. Adsorption Energy and M-N(O) Bond Distance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tan, K.; Zuluaga, S.; Wang, H.; Canepa, P.; Soliman, K.; Cure, J.; Li, J.; Thonhauser, T.; Chabal, Y.J. Interaction of Acid Gases SO2 and NO2 with Coordinatively Unsaturated Metal Organic Frameworks: M-MOF-74 (M = Zn, Mg, Ni, Co). Chem. Mater. 2017, 29, 4227–4235. [Google Scholar] [CrossRef]
- Khan, N.A.; Hasan, Z.; Jhung, S.H. Adsorptive removal of hazardous materials using metal-organic frameworks (MOFs): A review. J. Hazard. Mater. 2013, 244, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Hinks, N.J.; McKinlay, A.C.; Xiao, B.; Wheatley, P.S.; Morris, R.E. Metal organic frameworks as NO delivery materials for biological applications. Microporous Mesoporous Mater. 2010, 129, 330–334. [Google Scholar] [CrossRef]
- Bowser, B.; Brower, L.; Ohnsorg, M.; Gentry, L.; Beaudoin, C.; Anderson, M.; Bowser, B.H.; Brower, L.J.; Ohnsorg, M.L.; Gentry, L.K.; et al. Comparison of Surface-Bound and Free-Standing Variations of HKUST-1 MOFs: Effect of Activation and Ammonia Exposure on Morphology, Crystallinity, and Composition. Nanomaterials 2018, 8, 650. [Google Scholar] [CrossRef] [PubMed]
- Yepez, R.; García, S.; Schachat, P.; Sánchez-Sánchez, M.; González-Estefan, J.H.; González-Zamora, E.; Ibarra, I.A.; Aguilar-Pliego, J. Catalytic activity of HKUST-1 in the oxidation of trans-ferulic acid to vanillin. New J. Chem. 2015, 39, 5112–5115. [Google Scholar] [CrossRef]
- Campbell, J.; Székely, G.; Davies, R.P.; Braddock, D.C.; Livingston, A.G. Fabrication of hybrid polymer/metal organic framework membranes: Mixed matrix membranes versus in situ growth. J. Mater. Chem. A 2014, 2, 9260–9271. [Google Scholar] [CrossRef]
- Campbell, J.; Burgal, J.D.S.; Szekely, G.; Davies, R.P.; Braddock, D.C.; Livingston, A. Hybrid polymer/MOF membranes for Organic Solvent Nanofiltration (OSN): Chemical modification and the quest for perfection. J. Membr. Sci. 2016, 503, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Mosleh, S.; Rahimi, M.R.; Ghaedi, M.; Dashtian, K. HKUST-1-MOF–BiVO4 hybrid as a new sonophotocatalyst for simultaneous degradation of disulfine blue and rose bengal dyes: Optimization and statistical modelling. RSC Adv. 2016, 6, 61516–61527. [Google Scholar] [CrossRef]
- Razali, M.; Kim, J.F.; Attfield, M.; Budd, P.M.; Drioli, E.; Lee, Y.M.; Szekely, G. Sustainable wastewater treatment and recycling in membrane manufacturing. Green Chem. 2015, 17, 5196–5205. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, J.R.; Sánchez-González, E.; Pérez, E.; Schneider-Revueltas, E.; Martínez, A.; Tejeda-Cruz, A.; Islas-Jácome, A.; González-Zamora, E.; Ibarra, I.A. Structure stability of HKUST-1 towards water and ethanol and their effect on its CO2 capture properties. Dalton Trans. 2017, 46, 9192–9200. [Google Scholar] [CrossRef] [PubMed]
- Supronowicz, B.; Mavrandonakis, A.; Heine, T. Interaction of Biologically Important Organic Molecules with the Unsaturated Copper Centers of the HKUST-1 Metal−Organic Framework: An Ab-Initio Study. J. Phys. Chem. C 2015, 119, 3024–3032. [Google Scholar] [CrossRef]
- Khan, N.A.; Jhung, S.H. Adsorptive removal and separation of chemicals with metal-organic frameworks: Contribution of π-complexation. J. Hazard. Mater. 2017, 325, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wheatley, P.S.; Zhao, X.; Fletcher, A.J.; Fox, S.; Rossi, A.G.; Megson, I.L.; Bordiga, S.; Regli, L.; Thomas, K.M.; et al. High-Capacity Hydrogen and Nitric Oxide Adsorption and Storage in a Metal−Organic Framework. J. Am. Chem. Soc. 2007, 129, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, B.; Petit, C.; Bandosz, T.J. Reactive Adsorption of NO2 on Copper-Based Metal−Organic Framework and Graphite Oxide/Metal−Organic Framework Composites. ACS Appl. Mater. Interfaces 2010, 2, 3606–3613. [Google Scholar] [CrossRef] [PubMed]
- Borfecchia, E.; Maurelli, S.; Gianolio, D.; Groppo, E.; Chiesa, M.; Bonino, F.; Lamberti, C. Insights into Adsorption of NH3 on HKUST-1 Metal-Organic Framework: A Multitechnique Approach. J. Phys. Chem. C 2012, 116, 19839–19850. [Google Scholar] [CrossRef]
- Murray, L.J.; Dinca, M.; Yano, J.; Chavan, S.; Bordiga, S.; Brown, C.M.; Long, J.R. Highly-Selective and Reversible O2 Binding in Cr3(1,3,5-benzenetricarboxylate)2. J. Am. Chem. Soc. 2010, 132, 7856–7857. [Google Scholar] [CrossRef] [PubMed]
- Maniam, P.; Stock, N.; Chemie, A.; Kiel, D. Investigation of Porous Ni-Based Metal À Organic Frameworks Containing Paddle-Wheel Type Inorganic Building Units via High-Throughput Methods. Inorg. Chem. 2011, 50, 5085–5097. [Google Scholar] [CrossRef] [PubMed]
- Feldblyum, J.I.; Liu, M.; Gidley, D.W.; Matzger, A.J. Reconciling the Discrepancies between Crystallographic Porosity and Guest Access as Exemplified by Zn-HKUST-1. J. Am. Chem. Soc. 2011, 133, 18257–18263. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Schwarz, U.; Kaskel, S. Synthesis and properties of the metal-organic framework Mo3(BTC)2. J. Mater. Chem. 2006, 16, 2245–2248. [Google Scholar] [CrossRef]
- Howe, J.D.; Liu, Y.; Flores, L.; Dixon, D.A.; Sholl, D.S. Acid Gas Adsorption on Metal-Organic Framework Nanosheets as a Model of an “All-Surface” Material. J. Chem. Theory Comput. 2017, 13, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yang, L.; Zhang, Y.; Shi, Q.; Lu, R.; Deng, W. Accurate van der Waals Force Field for Gas Adsorption in Porous Materials. J. Comput. Chem. 2017, 38, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Ongari, D.; Tiana, D.; Stoneburner, S.J.; Gagliardi, L.; Smit, B. Origin of the Strong Interaction between Polar Molecules and Copper(II) Paddle-Wheels in Metal Organic Frameworks. J. Phys. Chem. C 2017, 121, 15135–15144. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ling, L.; Wang, B.; Fan, H.; Shangguan, J.; Mi, J. Adsorptive desulfurization with metal-organic frameworks: A density functional theory investigation. Appl. Surf. Sci. 2016, 387, 483–490. [Google Scholar] [CrossRef]
- Tan, K.; Zuluaga, S.; Gong, Q.; Gao, Y.; Nijem, N.; Li, J.; Thonhauser, T.; Chabal, Y.J. Competitive Coadsorption of CO2 with H2O, NH3, SO2, NO, NO2, N2, O2, and CH4 in M-MOF-74 (M = Mg, Co, Ni): The Role of Hydrogen Bonding. Chem. Mater. 2015, 27, 2203–2217. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, F.; Hu, J.; Zhao, S.; Liu, H.; Hu, Y. Screening of desulfurization adsorbent in metal–organic frameworks: A classical density functional approach. Chem. Eng. Sci. 2015, 137, 170–177. [Google Scholar] [CrossRef]
- Koh, H.S.; Rana, M.K.; Wong-Foy, A.G.; Siegel, D.J. Predicting Methane Storage in Open-Metal-Site Metal-Organic Frameworks. J. Phys. Chem. C 2015, 119, 13451–13458. [Google Scholar] [CrossRef]
- Fang, H.; Awati, R.; Boulfelfel, S.E.; Ravikovitch, P.I.; Sholl, D.S. First-Principles-Derived Force Fields for CH4 Adsorption and Diffusion in Siliceous Zeolites. J. Phys. Chem. C 2018, 122, 12880–12891. [Google Scholar] [CrossRef]
- Ketrat, S.; Maihom, T.; Wannakao, S.; Probst, M.; Nokbin, S.; Limtrakul, J. Coordinatively Unsaturated Metal–Organic Frameworks M3(btc)2 (M = Cr, Fe, Co, Ni, Cu, and Zn) Catalyzing the Oxidation of CO by N2O: Insight from DFT Calculations. Inorg. Chem. 2017, 56, 14005–14012. [Google Scholar] [CrossRef] [PubMed]
- Demir, H.; Greathouse, J.A.; Staiger, C.L.; Perry, J.J.; Allendorf, M.D.; Sholl, D.S. DFT-based force field development for noble gas adsorption in metal organic frameworks. J. Mater. Chem. A 2015, 3, 23539–23548. [Google Scholar] [CrossRef]
- Gu, Z.-G.; Heinke, L.; Wöll, C.; Neumann, T.; Wenzel, W.; Li, Q.; Fink, K.; Gordan, O.D.; Zahn, D.R.T. Experimental and theoretical investigations of the electronic band structure of metal-organic frameworks of HKUST-1 type. Appl. Phys. Lett. 2015, 107, 183301. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Luo, D.; Lin, Z.; Thiele, G.; Dehnen, S. Crystalline beryllium carboxylate frameworks containing inorganic chains of BeO4 tetrahedra. CrystEngComm 2013, 15, 1845. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab-initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.S.; Rana, M.K.; Hwang, J.; Siegel, D.J. Thermodynamic screening of metal-substituted MOFs for carbon capture. Phys. Chem. Chem. Phys. 2013, 15, 4573–4581. [Google Scholar] [CrossRef] [PubMed]
- Supronowicz, B.; Mavrandonakis, A.; Heine, T. Interaction of Small Gases with the Unsaturated Metal Centers of the HKUST-1 Metal Organic Framework. J. Phys. Chem. C 2013, 117, 14570–14578. [Google Scholar] [CrossRef]
- Nie, X.; Kulkarni, A.; Sholl, D.S. Computational Prediction of Metal Organic Frameworks Suitable for Molecular Infiltration as a Route to Development of Conductive Materials. J. Phys. Chem. Lett. 2015, 6, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Sholl, D.S.; Steckel, J.A. Density Functional Theory: A Practical Introduction; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Shi, X.R.; Wang, J.G.; Hermann, K. CO and NO Adsorption and Dissociation at the β-Mo2C(0001) Surface: A Density Functional Theory Study. J. Phys. Chem. C 2010, 114, 13630–13641. [Google Scholar] [CrossRef]
- Shi, X.-R.; Wang, S.-G.; Wang, J. Chemisorption of oxygen and subsequent reactions on low index surfaces of β–Mo2C: Insights from first-principles thermodynamics and kinetics. J. Mol. Catal. A Chem. 2016, 417, 53–63. [Google Scholar] [CrossRef]
- Khan, A.H.; Peikert, K.; Fröba, M.; Bertmer, M. NO adsorption in amino-modified Cu3(btc)2-type MOFs studied by solid-state NMR. Microporous Mesoporous Mater. 2015, 216, 111–117. [Google Scholar] [CrossRef]
- Petit, C.; Levasseur, B.; Mendoza, B.; Bandosz, T.J. Reactive adsorption of acidic gases on MOF/graphite oxide composites. Microporous Mesoporous Mater. 2012, 154, 107–112. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.-R.; Sholl, D.S. Nucleation of Rhn (n = 1–5) Clusters on γ-Al2O3 Surfaces: A Density Functional Theory Study. J. Phys. Chem. C 2012, 116, 10623–10631. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Contreras-García, J. Revealing non-covalent interactions in solids: NCI plots revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO-t1 | NO-t2 | NH3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| d(M-N) | d(N-O) | Ang. | ∆Eads | d(M-O) | d(N-O) | Ang. | ∆Eads | d(M-N) | ∆Eads | |
| gas | -- | 1.17 | -- | -- | -- | -- | -- | -- | -- | -- |
| Be | 1.96 | 1.17 | 174 | −20.0 | 2.23 | 1.17 | 168 | −0.4 | 1.75 | −136.7 |
| Fe | 1.71 | 1.18 | 178 | −81.6 | 1.91 | 1.19 | 178 | −15.0 | 2.11 | −60.3 |
| Ni | 1.81 | 1.18 | 122 | −157.0 | 3.30 | 1.17 | 140 | −3.4 | 2.07 | −95.2 |
| Cr | 1.83 | 1.19 | 151 | −41.0 | 3.24 | 1.17 | 175 | −0.9 | 2.37 | −35.0 |
| Co | 1.80 | 1.18 | 128 | −143.6 | 1.98 | 1.18 | 130 | −74.1 | 2.04 | −104.3 |
| Cu | 1.97 | 1.17 | 126 | −26.8(−21.31) 1 | 3.07 | 1.16 | 157 | −11.1 | 2.20 | −79.8 |
| V | 1.75 | 1.20 | 176 | −142.5 | 2.00 | 1.20 | 152 | −62.0 | 2.24 | −80.5 |
| Zn | 2.21 | 1.17 | 129 | −37.2 | 2.50 | 1.17 | 179 | −12.3 | 2.06 | −128.7 |
| Mo | 2.39 | 1.19 | 127 | −22.4 | 3.11 | 1.18 | 177 | −0.8 | 3.19 | −9.8 |
| Mn | 1.66 | 1.18 | 179 | −196.5 | 1.81 | 1.19 | 178 | −67.3 | 2.32 | −51.1 |
| W | 1.90 | 1.20 | 174 | −57.6 | 3.01 | 1.19 | 177 | −2.4 | 3.11 | −11.9 |
| Sn | 4.11 | 1.17 | 173 | −0.9 | 4.40 | 1.17 | 177 | −0.5 | 4.09 | −0.8 |
| Ti | 1.82 | 1.22 | 179 | −236.9 | 1.92 | 1.25 | 180 | −108.5 | 2.41 | −66.9 |
| Cd | 2.45 | 1.17 | 130 | −32.6 | 2.71 | 1.17 | 180 | −8.8 | 2.30 | −109.6 |
| Mg | 2.25 | 1.17 | 179 | −28.7 | 2.24 | 1.18 | 180 | −16.7 | 2.14 | −116.8 |
| Sc | 2.04 | 1.22 | 159 | −174.8 | 2.04 | 1.25 | 149 | −118.8 | 2.38 | −86.5 |
| Ca | 2.60 | 1.18 | 179 | −21.9 | 2.56 | 1.18 | 179 | −11.8 | 2.54 | −70.6 |
| Sr | 2.78 | 1.19 | 177 | −19.4 | 2.73 | 1.19 | 180 | −12.4 | 2.76 | −53.1 |
| Ba | 3.04 | 1.19 | 177 | −16.0 | 3.04 | 1.19 | 180 | −0.3 | 3.63 | −7.3 |
| (t1) | (t2) | (b1) | (b2) | |||||
|---|---|---|---|---|---|---|---|---|
| d(M-N) | ΔEads | d(M-O) | ΔEads | d(M-O/O’) | ΔEads | d(M-N/O) | ΔEads | |
| Be | 2.68 | −2.9 | 1.74 | −31.4 | b1 → t2 | -- | b2 → t2 | -- |
| Fe | 2.03 | −164.0 | 2.00 | −85.1 | b1 → t2 | -- | b2 → t1 | -- |
| Ni | 1.94 | −105.0 | 1.99 | −66.6 | b1 → t2 | -- | b2 → t1 | -- |
| Cr | t1 → b2 | -- | 2.03 | −64.5 | b1 → t2 | -- | 1.99/2.31 | −222.5 |
| Co | 1.95 | −115.9 | 1.97 | −82.1 | b1 → t2 | -- | b2 → t1 | -- |
| Cu | 2.22 | −13.2 | 2.24 | −40.8 | b1 → t2 | -- | b2 → t1 | -- |
| V | 2.15 | −82.2 | 1.89 | −116.5 | b1 → t2 | -- | 2.07/2.22 | −125.4 |
| Zn | 2.19 | −11.3 | 2.08 | −31.2 | b1 → t2 | -- | b2 → t2 | -- |
| Mo | 2.48 | −24.1 | 2.42 | −28.0 | b1 → t2 | -- | b2 → t1 | -- |
| Mn | 2.05 | −94.5 | 1.93 | −125.2 | b1 → t2 | -- | b2 → t1 | -- |
| W | 2.40 | −46.3 | 2.32 | −52.2 | b1 → t2 | -- | b2 → t1 | -- |
| Sn | 3.39 | −4.6 | 2.04 | −6.6 | 2.88/2.94 | −14.5 | b2 → t1 | -- |
| Ti | t1 → b2 | -- | t2 → b1 | -- | 2.20/2.19 | −224.0 | 2.05/1.99 | −245.5 |
| Cd | 2.43 | −11.5 | 2.41 | −27.9 | b1 → t2 | -- | b2 → t1 | -- |
| Mg | 2.28 | −5.7 | 2.08 | −40.6 | b1 → t2 | -- | b2 → t2 | -- |
| Sc | t1 → b2 | -- | 2.02 | −289.9 | 2.27/2.27 | −303.6 | 2.25/2.12 | −295.9 |
| Ca | t1 → t2 | -- | 2.43 | −42.1 | 2.63/2.62 | −32.9 | b2 → t2 | -- |
| Sr | t1 → t2 | -- | 2.60 | −43.8 | 2.75/2.86 | −40.0 | b2 → t2 | -- |
| Ba | 3.02 | −33.8 | 2.85 | −43.9 | 2.98/3.06 | −43.6 | b2 → t2 | -- |
| NO → N + O | NO2 → NO + O | NO2 → N + 2O | MH3 → NH2 + H | NH3 → NH + 2H | NH3 → N + 3H | |
|---|---|---|---|---|---|---|
| gas | 734.7 | 446.1 | 1180.8 | 471.2 | 887.6 | 1260.2 |
| Be | 619.3 | 329.3 | 948.6 | 284.1 2 | 691.8 2 | 1221.5 2 |
| Fe | 299.8 | 133.3 | 433.1 | 189.1 1 | 516.2 2 | 550.0 1 |
| Ni | 484.3 | 123.0 | 607.3 | 267.3 2 | 543.2 2 | 753.6 2 |
| Co | 397.4 | 99.9 | 497.3 | 264.4 1 | 542.3 1 | 739.1 1 |
| Cu | 586.9 | 318.0 | 904.9 | 279.9 2 | 472.7 2 | 662.9 2 |
| V | −139.2 | −174.9 | −314.1 | 200.7 2 | 353.1 2 | 506.6 2 |
| Sn | 336.8 | 132.5 | 469.3 | 268.2 2 | 581.8 2 | 946.5 2 |
| Ti | −147.1 | −162.4 | −309.5 | −2.9 1 | 100.3 1 | 316.5 1 |
| Cd | 649.8 | 339.7 | 989.5 | 351.0 2 | 667.4 2 | 927.1 2 |
| Sc | 51.8 | −36.7 | 15.1 | 153.8 2 | 570.6 2 | 1031.8 2 |
| Ba | 622.9 | 355.6 | 978.5 | 295.2 2 | 594.3 2 | 843.3 2 |
| Metal | qNO | qNO2-t1 | qNO2-t2 | qNO2-b1 | qNO2-b2 | qNH3 |
|---|---|---|---|---|---|---|
| Be | 0.01 | 0.00 | −0.20 | -- | -- | 0.11 |
| Fe | −0.10 | −0.45 | −0.55 | -- | -- | 0.19 |
| Ni | −0.15 | −0.26 | −0.38 | -- | -- | 0.17 |
| Cr | −0.28 | -- | −0.27 | -- | −0.40 | 0.08 |
| Co | −0.19 | −0.37 | −0.45 | -- | -- | 0.19 |
| Cu | 0.04 | −0.17 | −0.09 | -- | -- | 0.11 |
| V | −0.45 | −0.49 | −0.58 | -- | −0.53 | 0.14 |
| Zn | 0.04 | −0.05 | −0.15 | -- | -- | 0.18 |
| Mo | −0.19 | −0.42 | −0.44 | -- | -- | 0.03 |
| Mn | −0.33 | −0.51 | −0.57 | -- | -- | 0.13 |
| W | −0.19 | −0.56 | −0.60 | -- | -- | 0.04 |
| Sn | −0.01 | −0.08 | −0.01 | −0.29 | -- | 0.00 |
| Ti | −0.65 | -- | -- | −0.72 | −0.72 | 0.07 |
| Cd | −0.01 | −0.10 | −0.07 | -- | -- | 0.16 |
| Mg | −0.04 | −0.32 | −0.16 | -- | -- | 0.06 |
| Sc | −0.65 | -- | −0.75 | −0.77 | −0.73 | 0.08 |
| Ca | −0.09 | -- | −0.31 | −0.44 | -- | 0.06 |
| Sr | −0.19 | -- | −0.48 | −0.57 | -- | 0.04 |
| Ba | −0.22 | −0.52 | −0.52 | −0.57 | -- | 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, S.; Zhang, Y.; Lu, N.; Ma, P.; Wang, J.; Shi, X.-R. A DFT Screening of M-HKUST-1 MOFs for Nitrogen-Containing Compounds Adsorption. Nanomaterials 2018, 8, 958. https://doi.org/10.3390/nano8110958
Zong S, Zhang Y, Lu N, Ma P, Wang J, Shi X-R. A DFT Screening of M-HKUST-1 MOFs for Nitrogen-Containing Compounds Adsorption. Nanomaterials. 2018; 8(11):958. https://doi.org/10.3390/nano8110958
Chicago/Turabian StyleZong, Shibiao, Yajing Zhang, Na Lu, Pan Ma, Jianguo Wang, and Xue-Rong Shi. 2018. "A DFT Screening of M-HKUST-1 MOFs for Nitrogen-Containing Compounds Adsorption" Nanomaterials 8, no. 11: 958. https://doi.org/10.3390/nano8110958