

Molecular Dynamics Study of Nanoribbon Formation by Encapsulating Cyclic Hydrocarbon Molecules inside Single-Walled Carbon Nanotube


Abstract
:1. Introduction
2. Methods
3. Results and Discussion
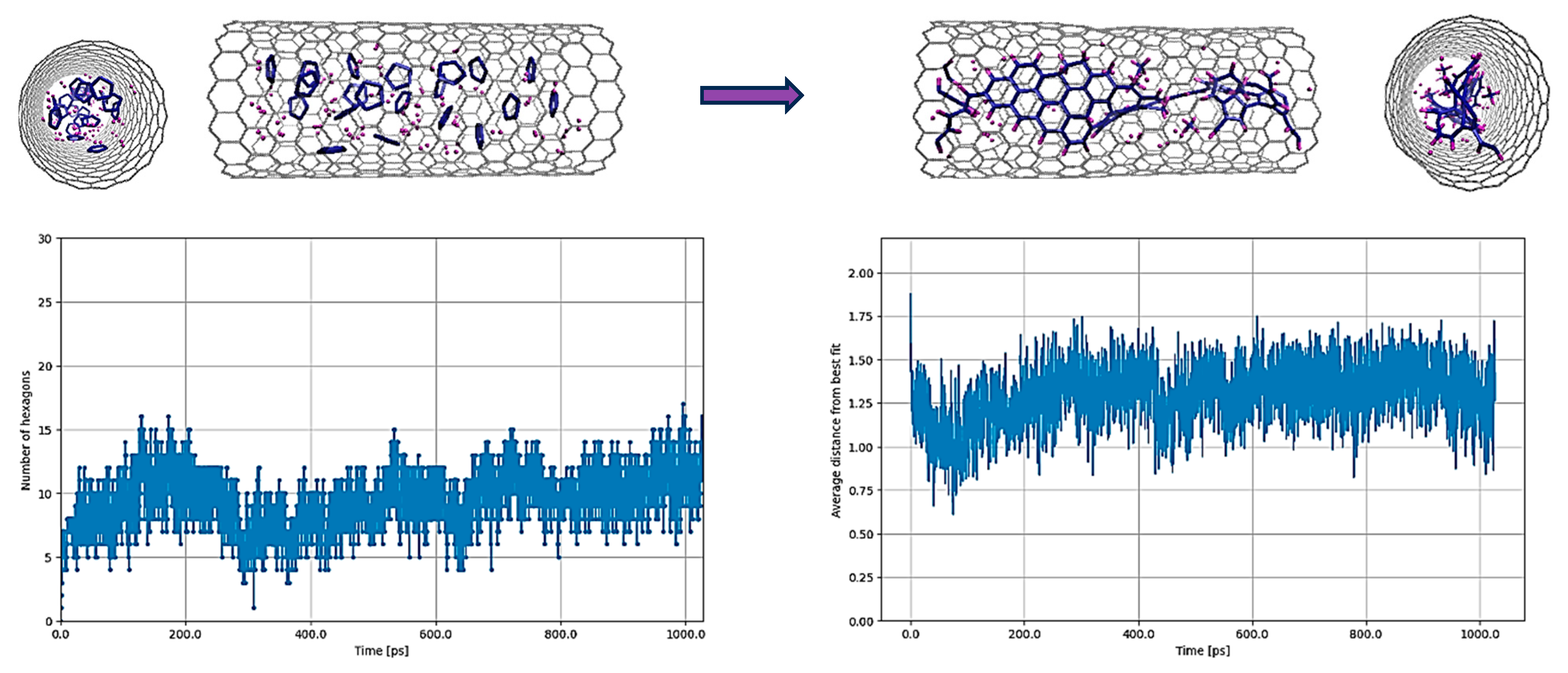
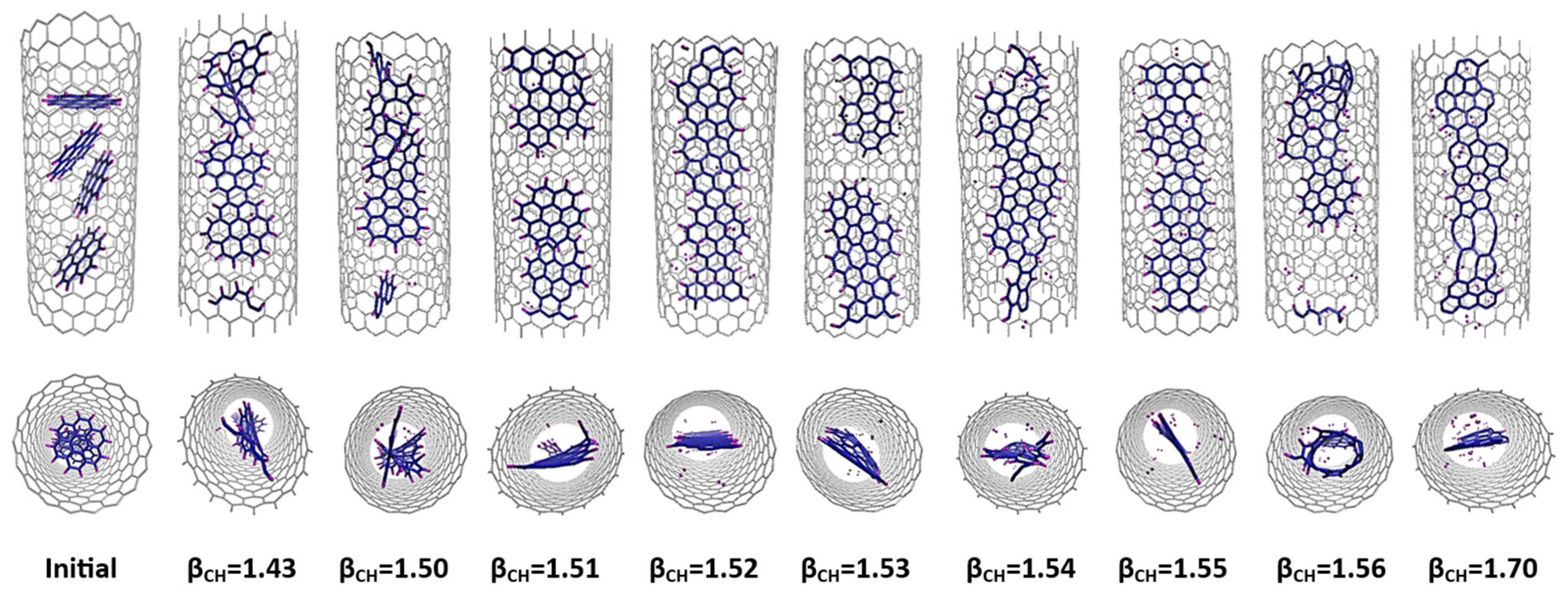
3.1. C5H5@SWCNT
3.2. C6H6@SWCNT
3.3. C20H12@SWCNT and C24H12@SWCNT
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, N.; Guan, L. A chemical combination reaction within single-walled carbon nanotubes. Nanoscale 2010, 2, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Khlobystov, A.N. Carbon nanotubes: From nano test tube to nano-reactor. ACS Nano 2011, 5, 9306–9312. [Google Scholar] [CrossRef] [PubMed]
- Miners, S.A.; Rance, G.A.; Khlobystov, A.N. Chemical reactions confined within carbon nanotubes. Chem. Soc. Rev. 2016, 45, 4727–4746. [Google Scholar] [CrossRef] [PubMed]
- Cadena, A.; Botka, B.; Kamarás, K. Organic molecules encapsulated in single-walled carbon nanotubes. Oxf. Open Mater. Sci. 2021, 1, itab009. [Google Scholar] [CrossRef]
- Hou, Z.; Yee, M. Electronic and transport properties of graphene nanoribbons. In Proceedings of the IEEE 2007 7th IEEE Conference on Nanotechnology (IEEE NANO), Hong Kong, China, 2–5 August 2007; pp. 554–557. [Google Scholar]
- Huang, B.; Yan, Q.M.; Li, Z.Y.; Duan, W.H. Towards graphene nanoribbon-based electronics. Front. Phys. China 2009, 4, 269–279. [Google Scholar] [CrossRef]
- Akhukov, M.A.; Yuan, S.; Fasolino, A.; Katsnelson, M.I. Electronic, magnetic and transport properties of graphene ribbons terminated by nanotubes. New J. Phys. 2012, 14, 123012. [Google Scholar] [CrossRef]
- Sun, J.; Li, Y.; Peng, Q.; Hou, S.; Zou, D.; Shang, Y.; Li, Y.; Li, P.; Du, Q.; Wang, Z.; et al. Macroscopic, flexible, high-performance graphene ribbons. ACS Nano 2013, 7, 10225–10232. [Google Scholar] [CrossRef] [PubMed]
- Celis, A.; Nair, M.N.; Taleb-Ibrahimi, A.; Conrad, E.H.; Berger, C.; De Heer, W.A.; Tejeda, A. Graphene nanoribbons: Fabrication, properties and devices. J. Phys. D Appl. Phys. 2016, 49, 143001. [Google Scholar] [CrossRef]
- Martini, L.; Chen, Z.; Mishra, N.; Barin, G.B.; Fantuzzi, P.; Ruffieux, P.; Fasel, R.; Feng, X.; Narita, A.; Coletti, C.; et al. Structure-dependent electrical properties of graphene nanoribbon devices with graphene electrodes. Carbon 2019, 146, 36–43. [Google Scholar] [CrossRef]
- Lukomskaya, M.V.; Pavlovsky, O.V. Electronic Properties of Graphene Nanoribbons. Phys. At. Nucl. 2020, 83, 1611–1614. [Google Scholar] [CrossRef]
- Houtsma, R.K.; de la Rie, J.; Stöhr, M. Atomically precise graphene nanoribbons: Interplay of structural and electronic properties. Chem. Soc. Rev. 2021, 50, 6541–6568. [Google Scholar] [CrossRef] [PubMed]
- Talyzin, A.V.; Anoshkin, I.V.; Krasheninnikov, A.V.; Nieminen, R.M.; Nasibulin, A.G.; Jiang, H.; Kauppinen, E.I. Synthesis of graphene nanoribbons encapsulated in single-walled carbon nanotubes. Nano Lett. 2011, 11, 4352–4356. [Google Scholar] [CrossRef] [PubMed]
- Kuzmany, H.; Shi, L.; Martinati, M.; Cambré, S.; Wenseleers, W.; Kürti, J.; Koltai, J.; Kukucska, G.; Cao, K.; Kaiser, U.; et al. Well-defined sub-nanometer graphene ribbons synthesized inside carbon nanotubes. Carbon 2021, 171, 221–229. [Google Scholar] [CrossRef]
- Cadena, A.; Botka, B.; Pekker, Á.; Tschannen, C.D.; Lombardo, C.; Novotny, L.; Khlobystov, A.N.; Kamarás, K. Molecular encapsulation from the liquid phase and graphene nanoribbon growth in carbon nanotubes. J. Phys. Chem. Lett. 2022, 13, 9752–9758. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Irle, S.; Morokuma, K. Formation mechanism of polycyclic aromatic hydrocarbons in benzene combustion: Quantum chemical molecular dynamics simulations. J. Chem. Phys. 2010, 132, 224303. [Google Scholar] [CrossRef]
- Saha, B.; Shindo, S.; Irle, S.; Morokuma, K. Quantum chemical molecular dynamics simulations of dynamic fullerene self-assembly in benzene combustion. ACS Nano 2009, 3, 2241–2257. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, X.; Qian, H.-J.; Ohta, Y.; Wu, X.; Eres, G.; Morokuma, K.; Irle, S. Quantum chemical simulations reveal acetylene-based growth mechanisms in the chemical vapor deposition synthesis of carbon nanotubes. Carbon 2014, 72, 22–37. [Google Scholar] [CrossRef]
- De Tomas, C.; Suarez-Martinez, I.; Marks, N.A. Graphitization of amorphous carbons: A comparative study of interatomic potentials. Carbon 2016, 109, 681–693. [Google Scholar] [CrossRef]
- De Tomas, C.; Aghajamali, A.; Jones, J.L.; Lim, D.J.; López, M.J.; Suarez-Martinez, I.; Marks, N.A. Transferability in interatomic potentials for carbon. Carbon 2019, 155, 624–634. [Google Scholar] [CrossRef]
- László, I.; Gyimesi, B.; Koltai, J.; Kürti, J. Molecular dynamics simulation of carbon structures inside small diameter carbon nanotubes. Phys. Status Solidi (b) 2017, 254, 1700206. [Google Scholar] [CrossRef]
- Eskandari, S.; Koltai, J.; László, I.; Vaezi, M.; Kürti, J. Formation of nanoribbons by carbon atoms confined in a single-walled carbon nanotube—A molecular dynamics study. J. Chem. Phys. 2023, 158, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Tersoff, J. Empirical interatomic potential for carbon, with applications to amorphous carbon. Phys. Rev. Lett. 1988, 61, 2879. [Google Scholar] [CrossRef] [PubMed]
- Tersoff, J. Modeling solid-state chemistry: Interatomic potentials for multicomponent systems. Phys. Rev. B 1989, 39, 5566. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783. [Google Scholar] [CrossRef]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Chenoweth, K.; Van Duin, A.C.; Persson, P.; Cheng, M.J.; Oxgaard, J.; Goddard Iii, W.A. Development and application of a ReaxFF reactive force field for oxidative dehydrogenation on vanadium oxide catalysts. J. Phys. Chem. C 2008, 112, 14645–14654. [Google Scholar] [CrossRef]
- Knippenberg, M.T.; Kum, O.; Stuart, S.J. Structural study of amorphous carbon using adaptive interatomic reactive empirical bond-order potential model. In Proceedings of the NSTI Nanotechnology Conference and Trade Show, Anaheim, CA, USA, 8–12 May 2005. [Google Scholar]
- O’connor, T.C.; Andzelm, J.; Robbins, M.O. AIREBO-M: A reactive model for hydrocarbons at extreme pressures. J. Chem. Phys. 2015, 142, 024903. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Luo, Y.-R.; Cheng, J.-P. Bond Dissociation Energies. In CRC Handbook of Chemistry and Physics, 96th ed.; CRC: Boca Raton, FL, USA, 2015. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor | Initial Number of C-C Bonds | Initial Number of C-H Bonds | Initial H/C Ratio | βC-H (Å−1) Ranges for GNR Formation | Average C-H Bond Energy (eV) | Final Number of C-C Bond | Final Number of C-H Bonds | Final H/C Ratio | Final Value of Nonplanarity (Å) |
|---|---|---|---|---|---|---|---|---|---|
| 20 C5H5 | 100 | 100 | 1 | 1.54–1.55 | 3.76 | 131 | 31 | 0.31 | 0.75 |
| 20C5 + 50H2 | 100 | 0 | 0 | 1.51–1.56 | 3.83 | 126 | 30 | 0.30 | 0.70 |
| 16 C6H6 | 96 | 96 | 1 | 1.53–1.56 | 3.73 | 131 | 29 | 0.30 | 0.70 |
| 16C6 + 48H2 | 96 | 0 | 0 | 1.51–1.54 | 3.89 | 124 | 29 | 0.30 | 0.82 |
| 5 C20H12 | 120 | 60 | 0.60 | 1.54–1.56 | 3.73 | 129 | 30 | 0.30 | 0.66 |
| 4 C24H12 | 120 | 48 | 0.50 | 1.51–1.55 | 3.86 | 124 | 25 | 0.26 | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eskandari, S.; Koltai, J.; László, I.; Kürti, J. Molecular Dynamics Study of Nanoribbon Formation by Encapsulating Cyclic Hydrocarbon Molecules inside Single-Walled Carbon Nanotube. Nanomaterials 2024, 14, 627. https://doi.org/10.3390/nano14070627
Eskandari S, Koltai J, László I, Kürti J. Molecular Dynamics Study of Nanoribbon Formation by Encapsulating Cyclic Hydrocarbon Molecules inside Single-Walled Carbon Nanotube. Nanomaterials. 2024; 14(7):627. https://doi.org/10.3390/nano14070627
Chicago/Turabian StyleEskandari, Somayeh, János Koltai, István László, and Jenő Kürti. 2024. "Molecular Dynamics Study of Nanoribbon Formation by Encapsulating Cyclic Hydrocarbon Molecules inside Single-Walled Carbon Nanotube" Nanomaterials 14, no. 7: 627. https://doi.org/10.3390/nano14070627