
Photocatalysis Based on Metal Halide Perovskites for Organic Chemical Transformations


Abstract
:1. Introduction

2. Factors Effecting the Photocatalytic Performance
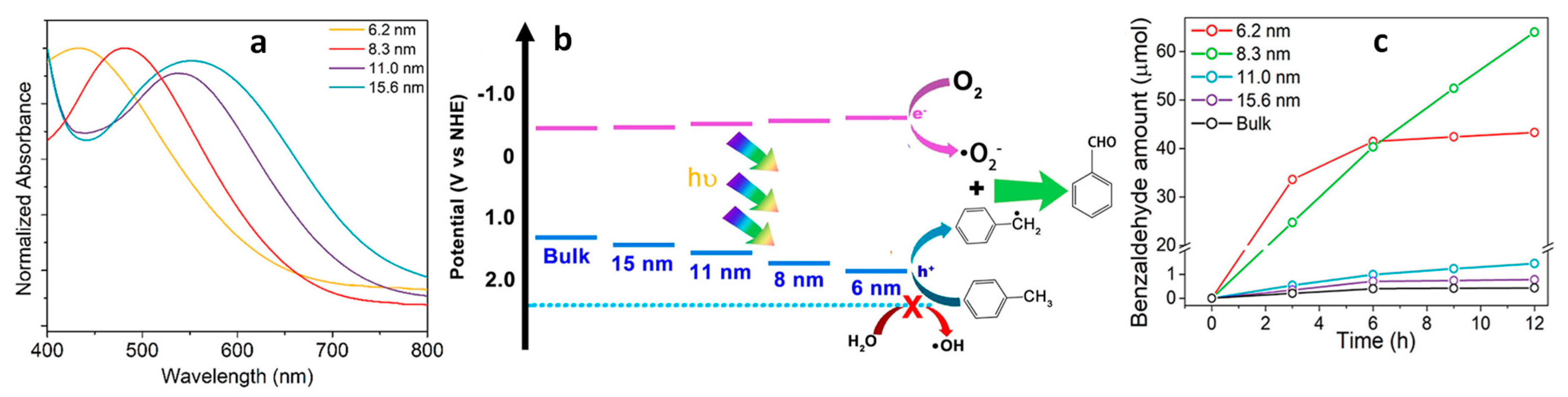
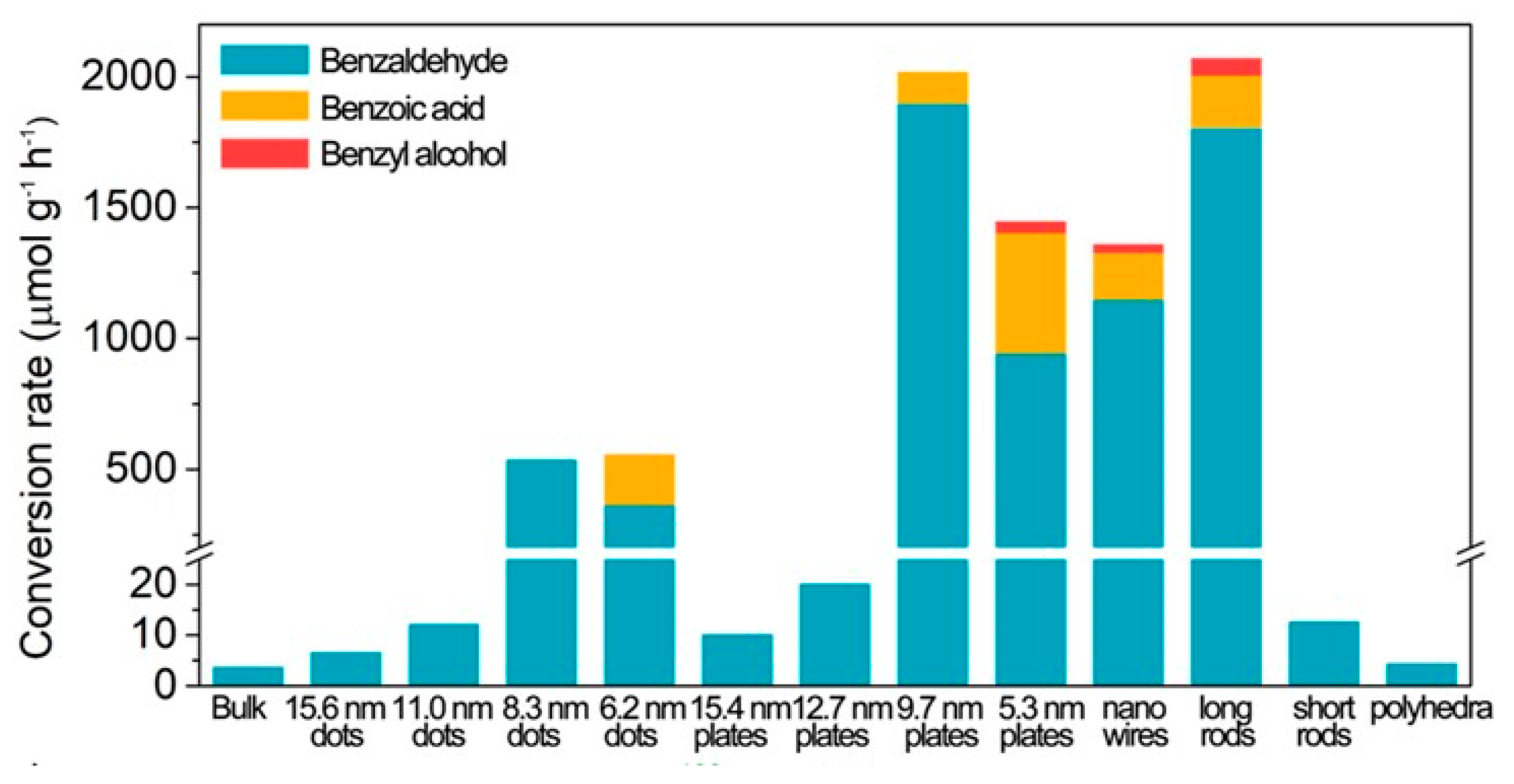
2.1. Effect of Size of NCs on Photocatalytic Activity
2.1.1. Size Dependent Photocatalytic Activity of Lead Halide PNCs
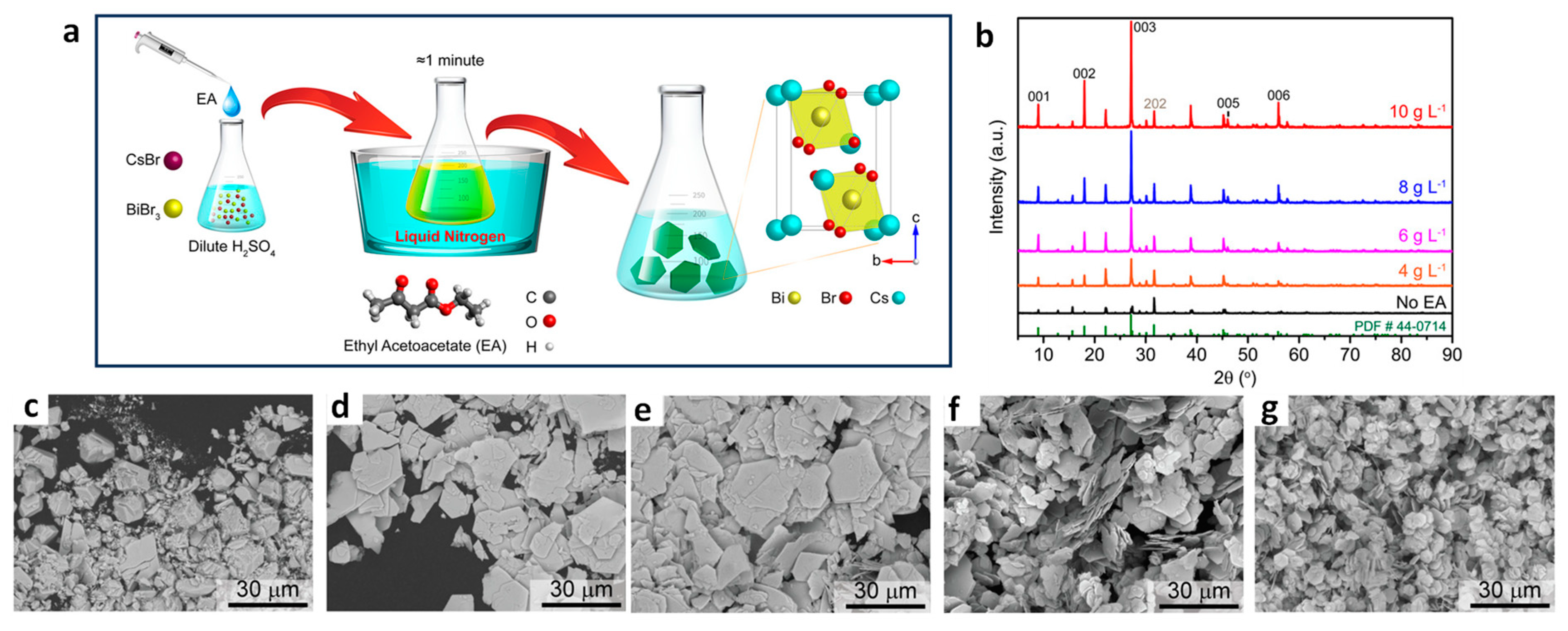
2.1.2. Size Dependent Photocatalytic Activity of Lead-Free Halide PNCs
2.2. Stability and Reaction Condition Tolerance
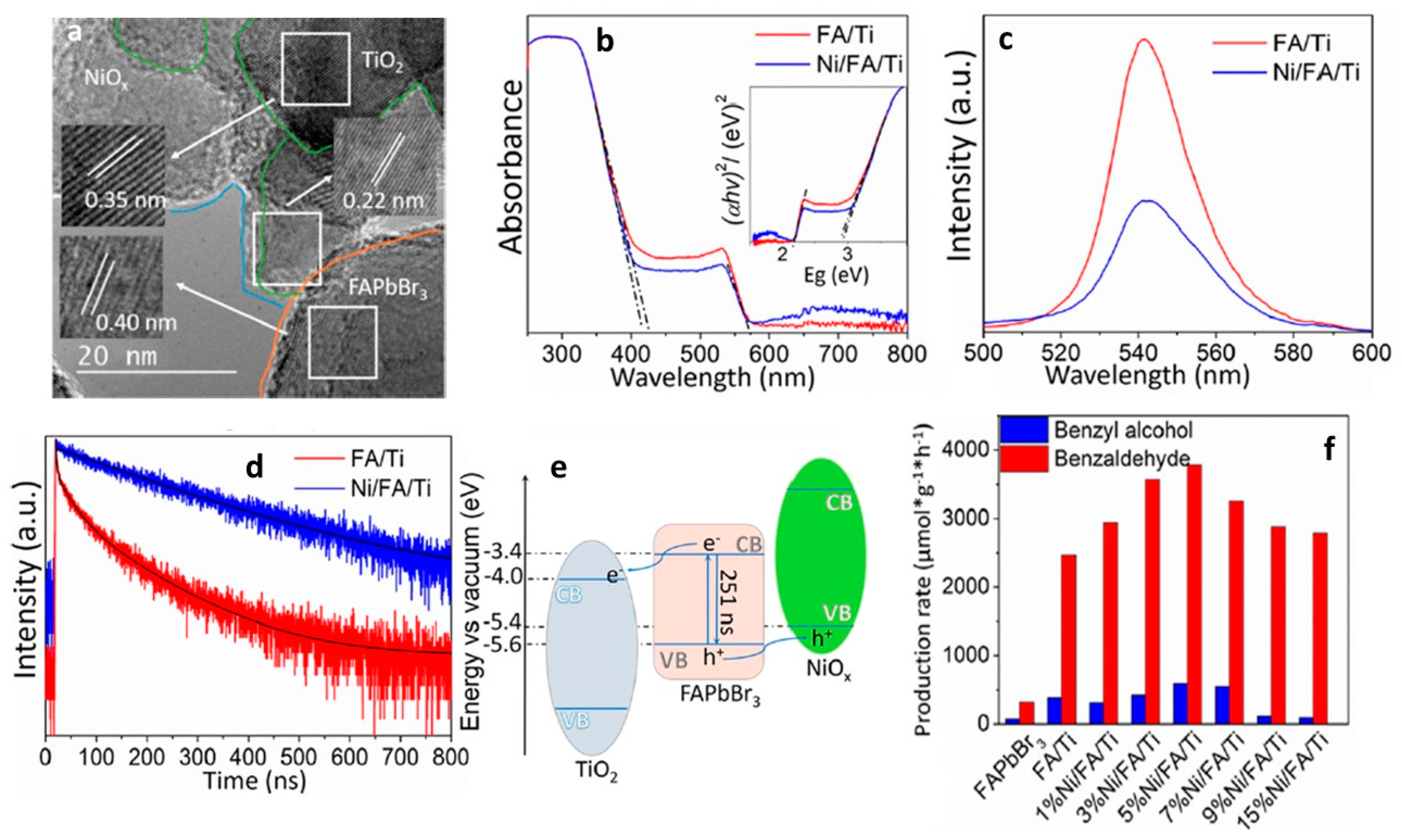
2.2.1. Role of a Co-Catalyst
2.2.2. Role of Acidity
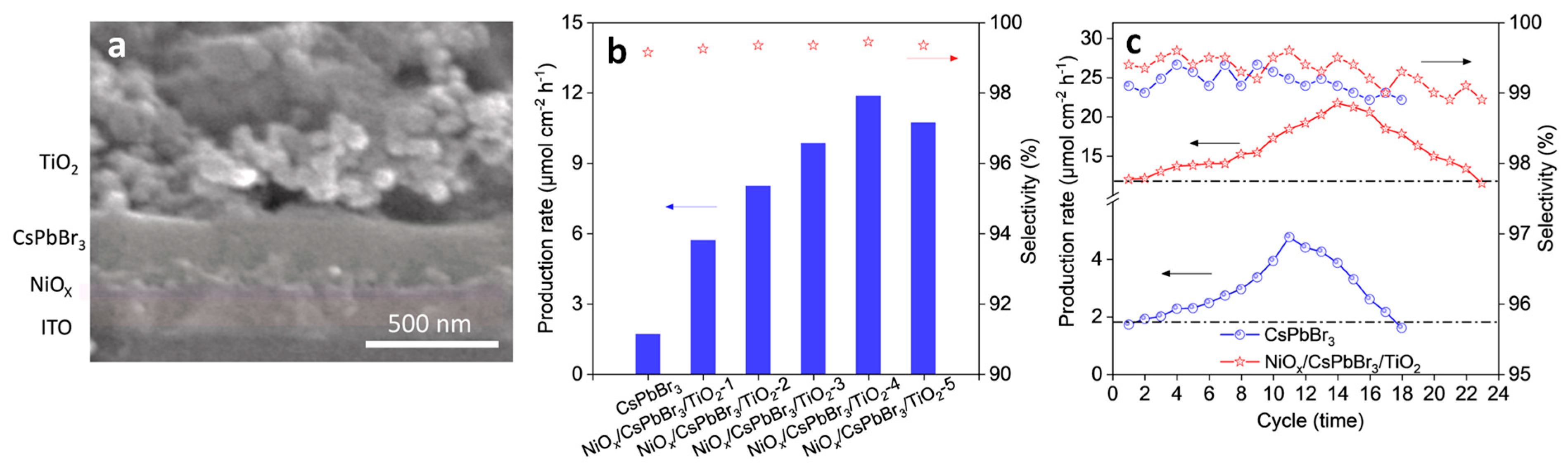
2.2.3. Role of Air
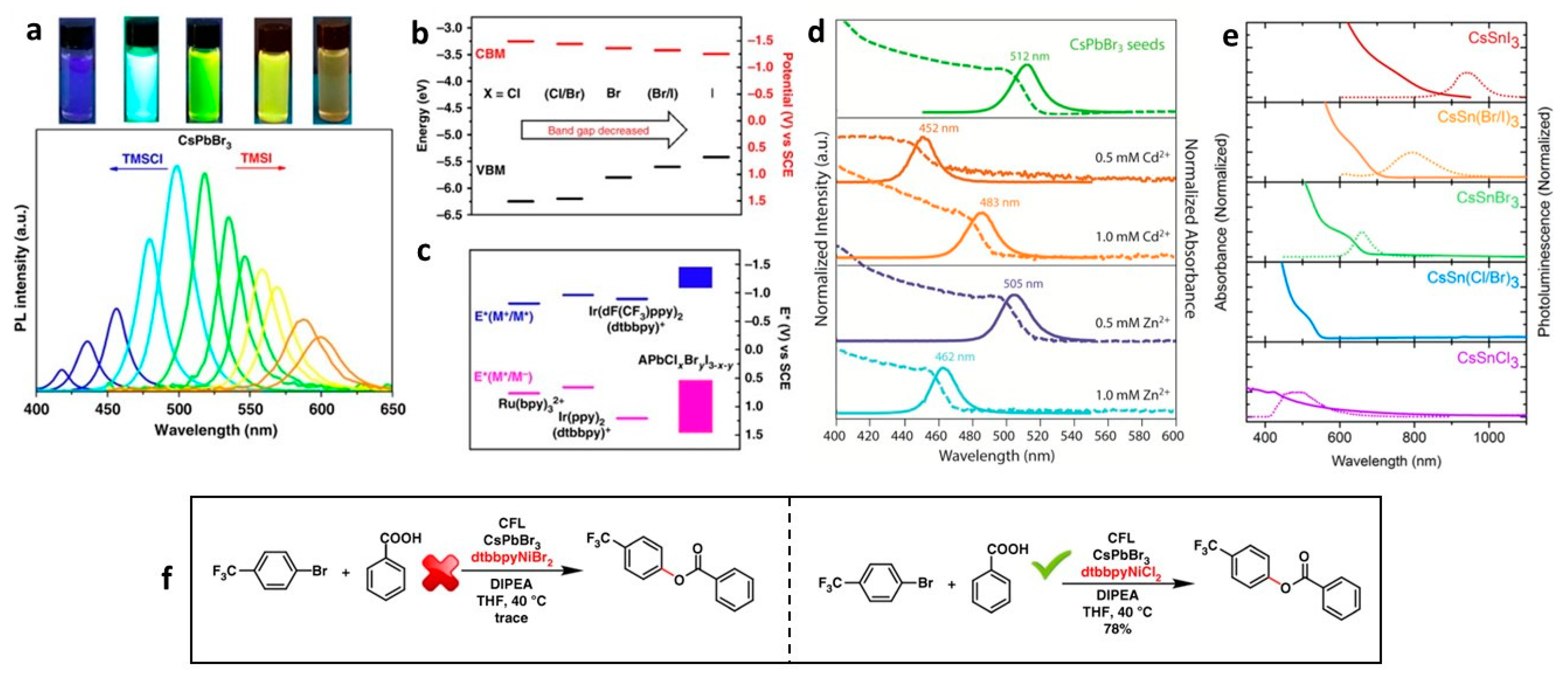
2.3. Role of Bandgap Tuning of Perovskite NCs on Photocatalytic Organic Reactions
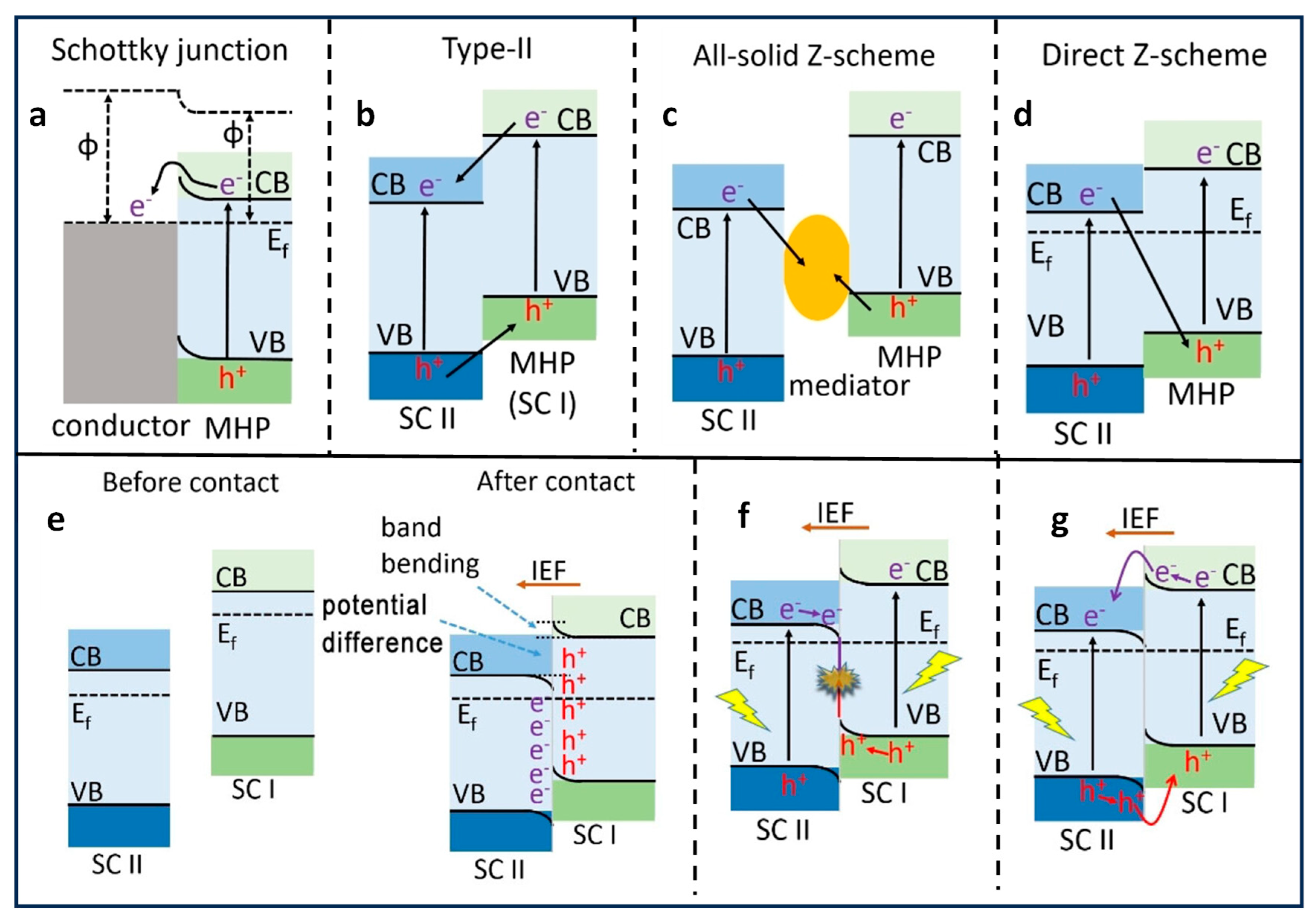
3. Types of Heterojunctions for Photocatalysis
4. Carbon–Carbon (C–C) Bond Formation
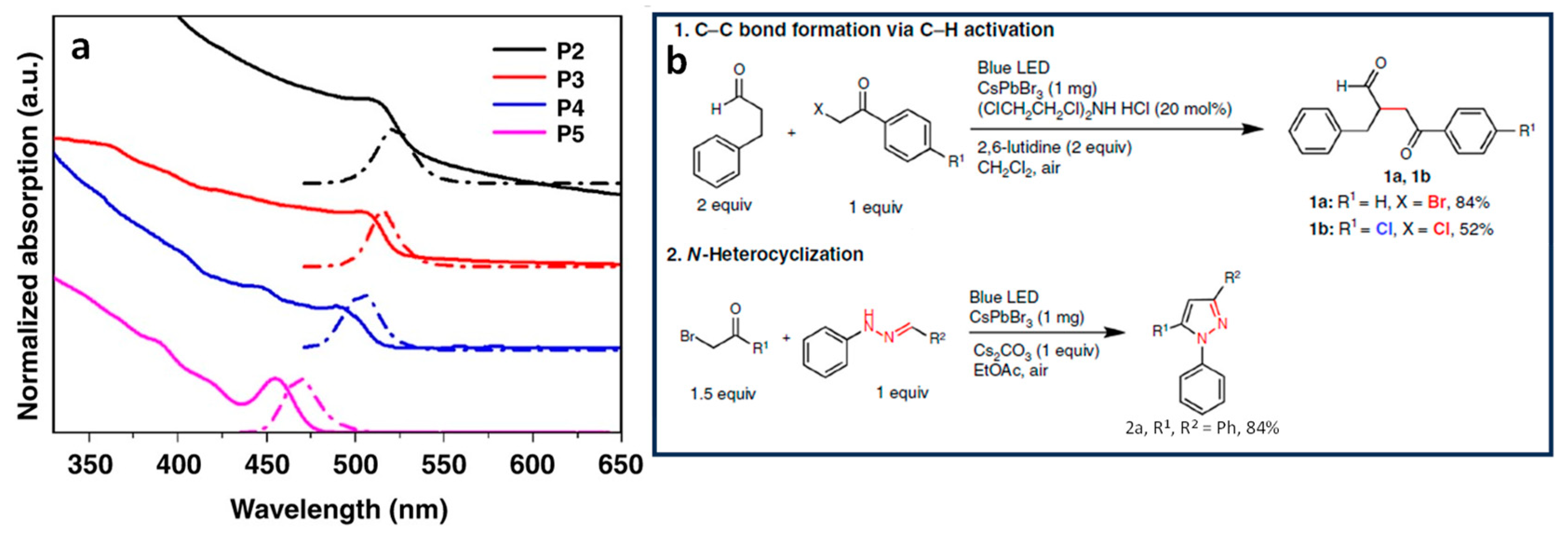
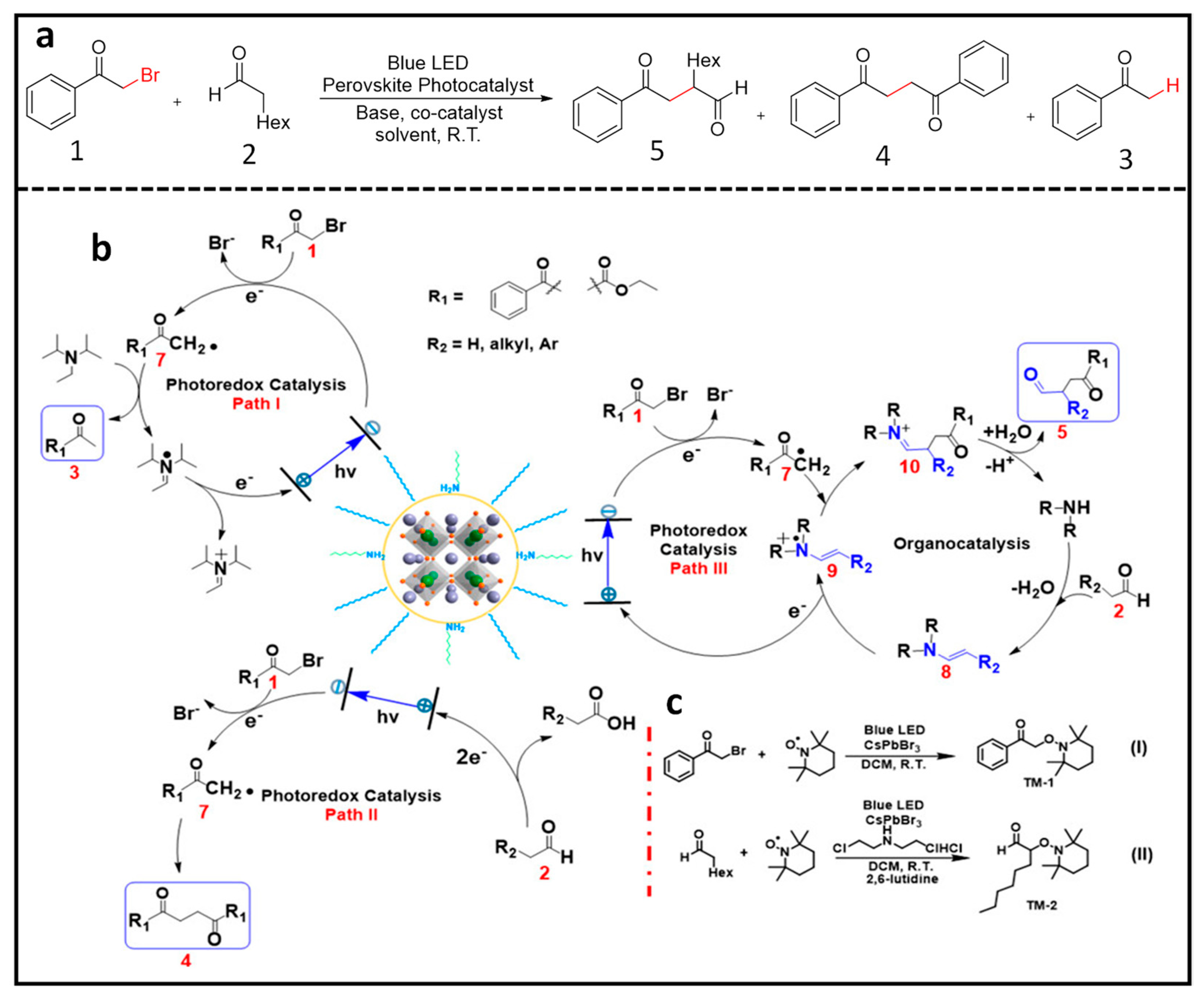
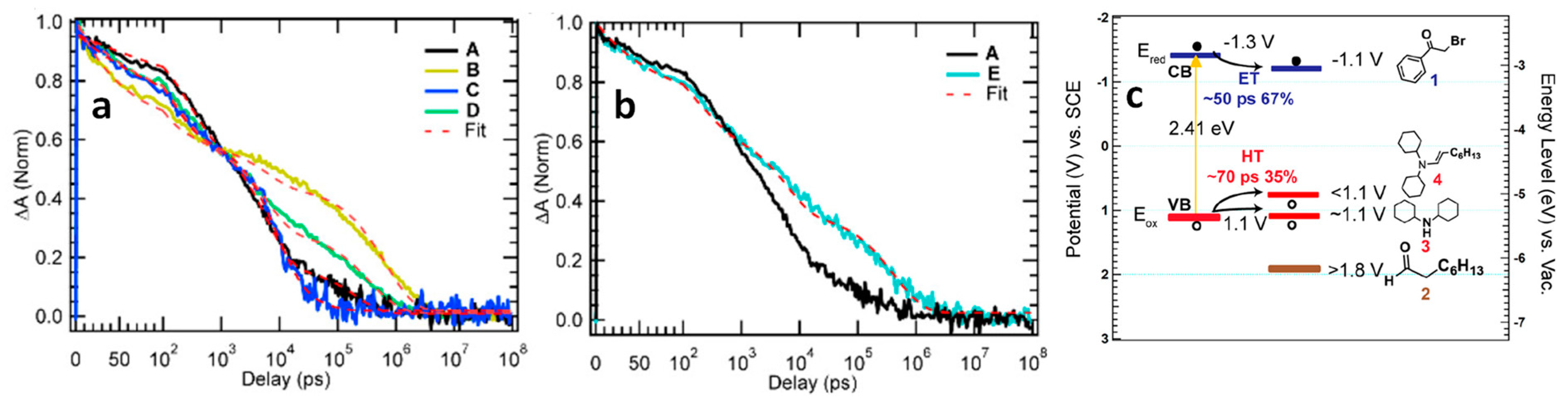
4.1. α-Alkylation of Aldehydes Using LHPNCs
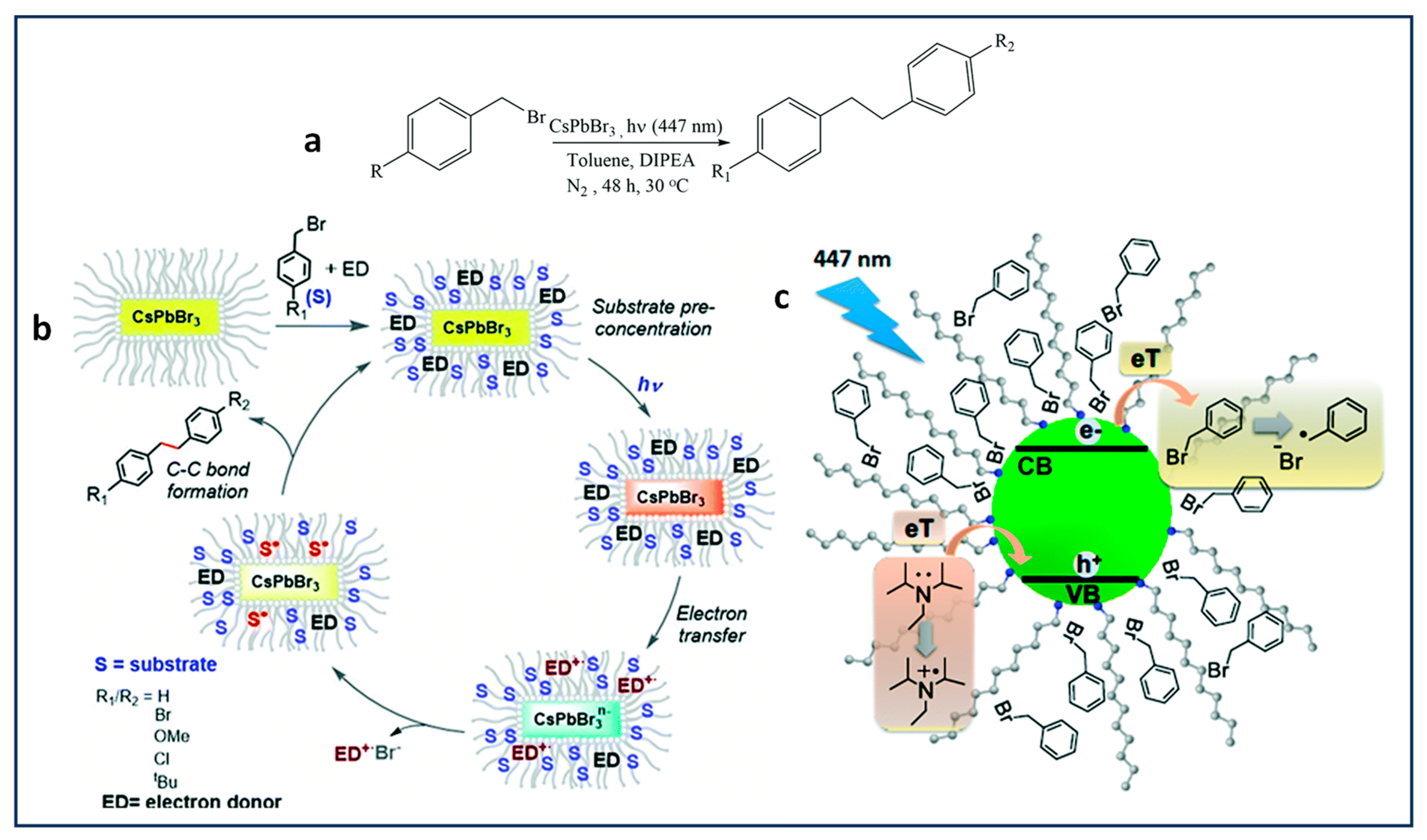
4.2. Benzyl Bromide Coupling Using LHP NCs
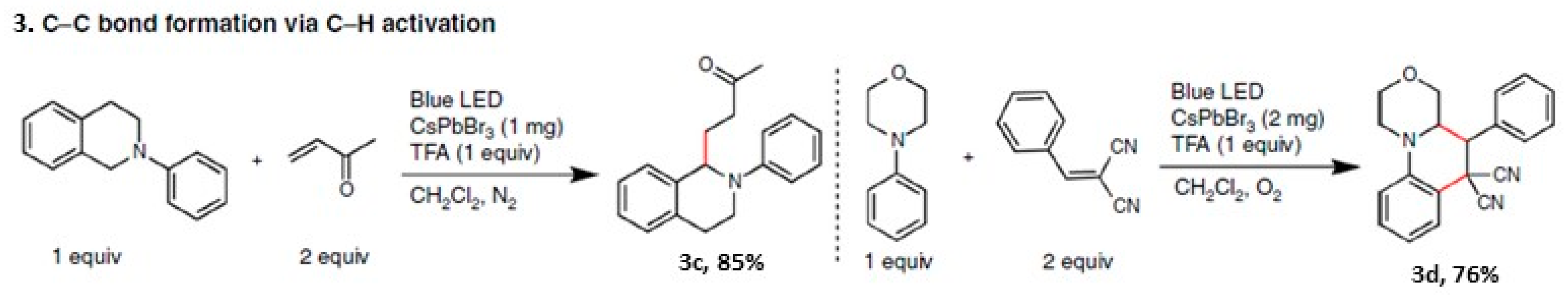
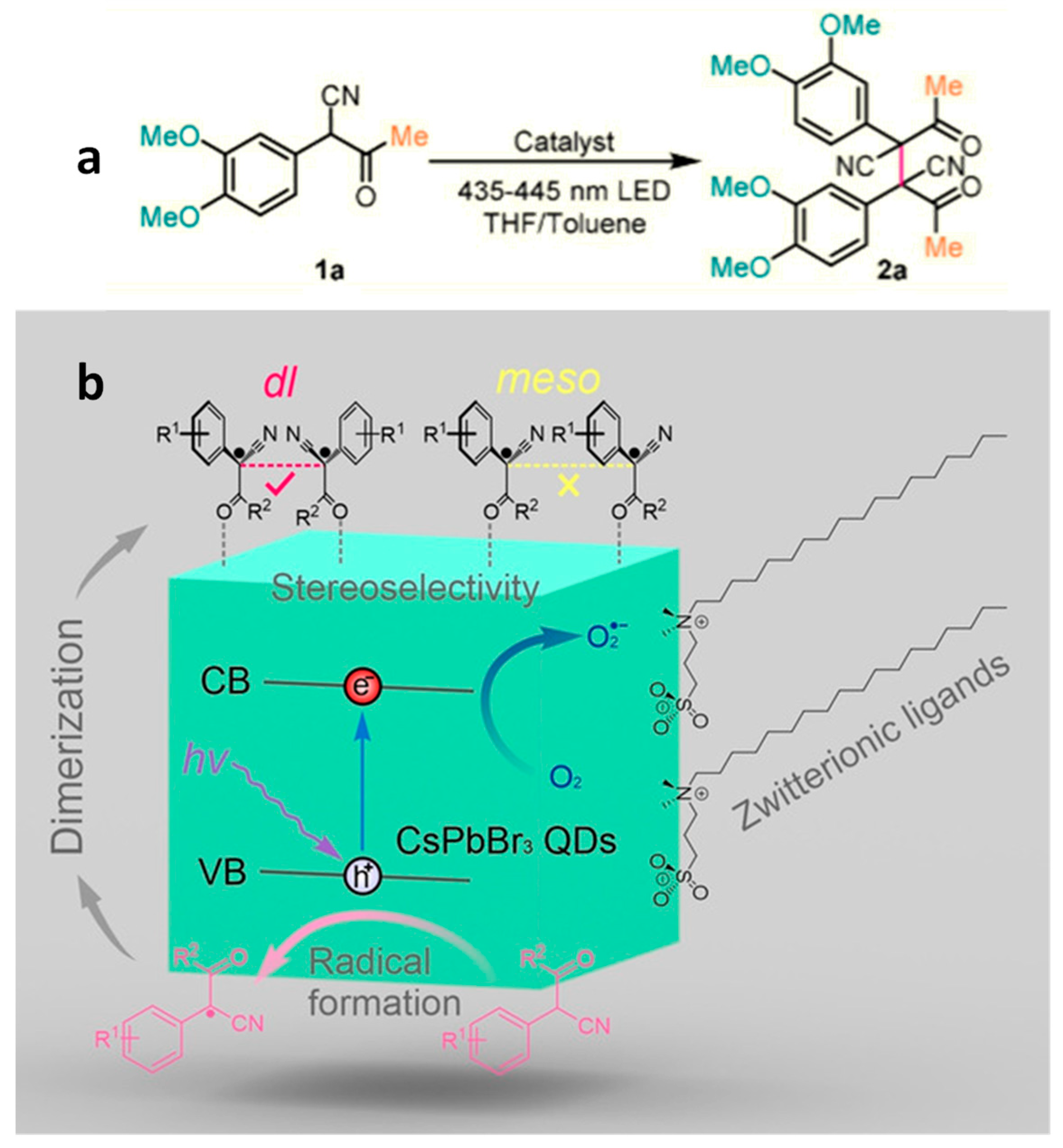
4.3. Stereo Selective C–C Oxidative Coupling Reaction
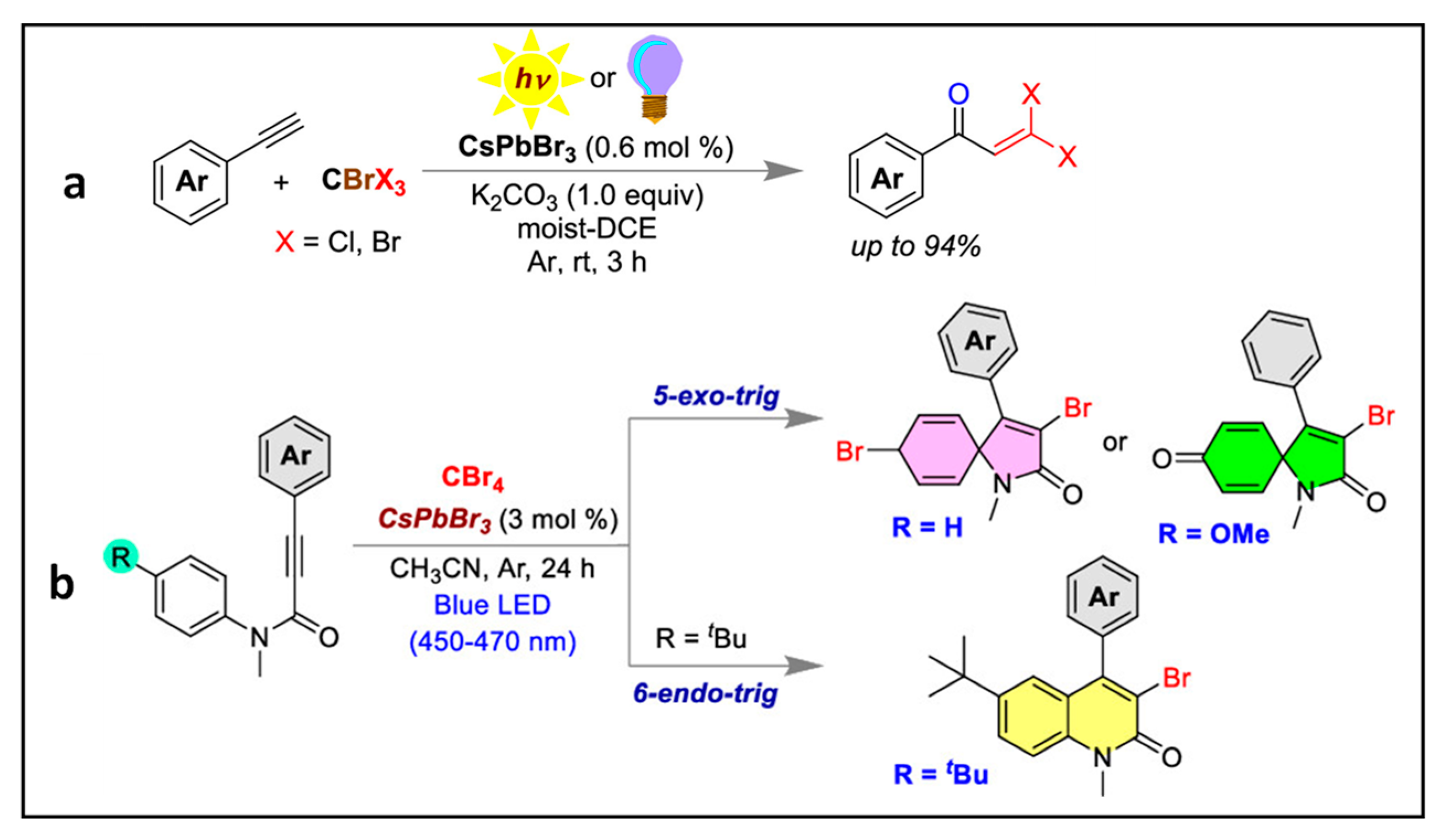
4.4. Synthesis of Gem-Dihaloenones by the Activation of C-Br Bonds of CBrX3 (X = Cl, Br)
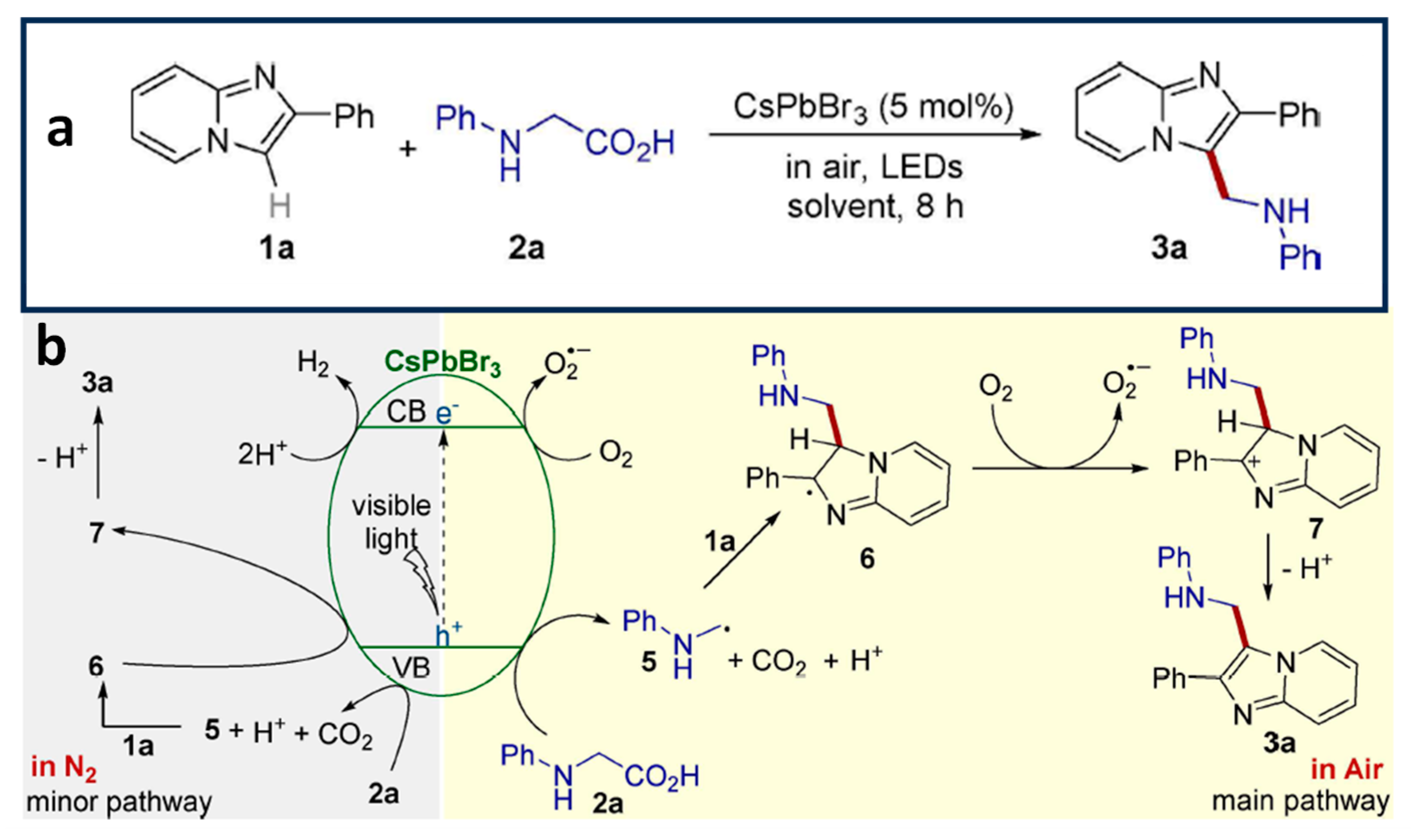
4.5. Aminomethylation of Imidazo-Fused Heterocycles
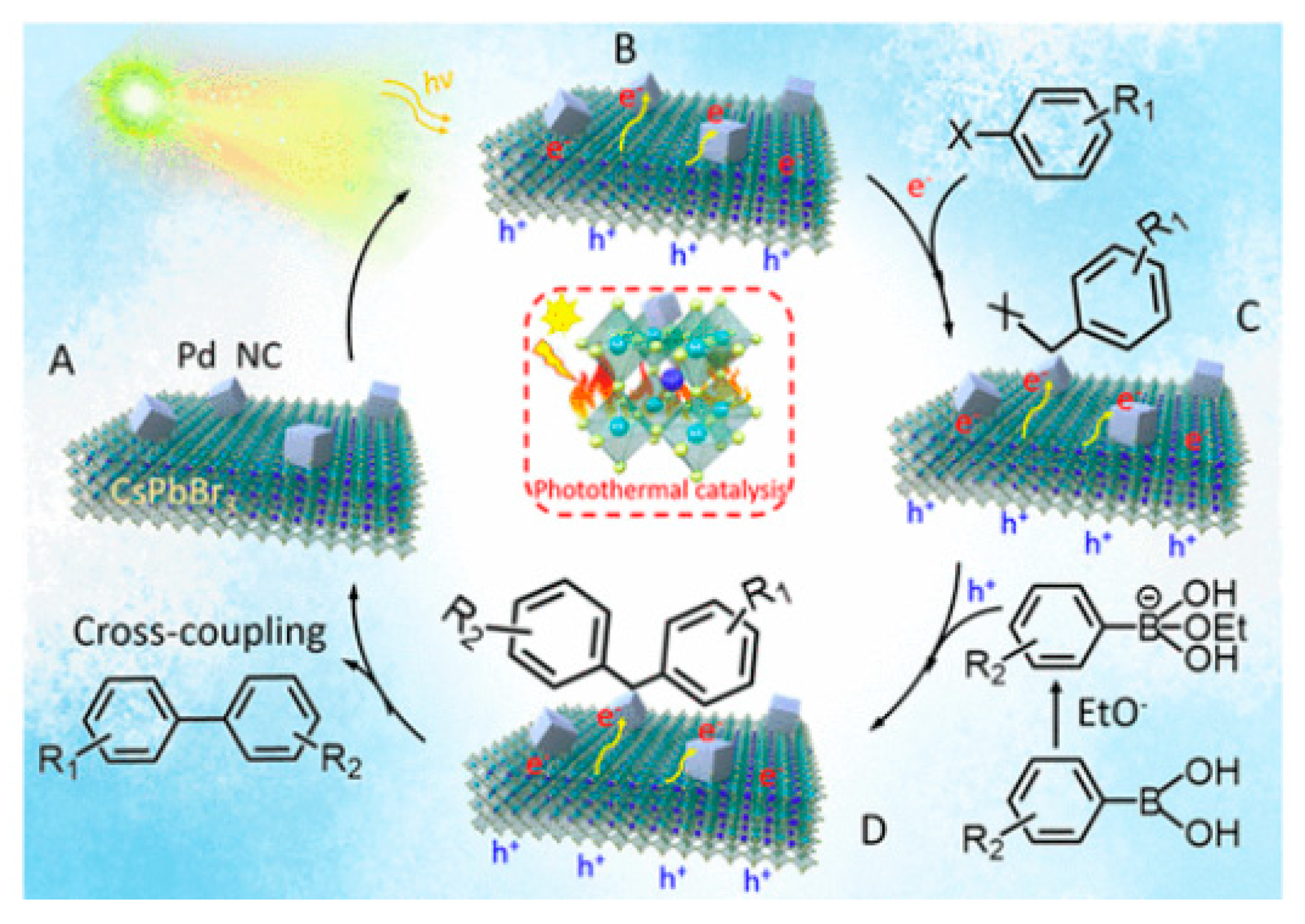
4.6. Photothermal Suzuki Coupling CsPbBr3/Pd
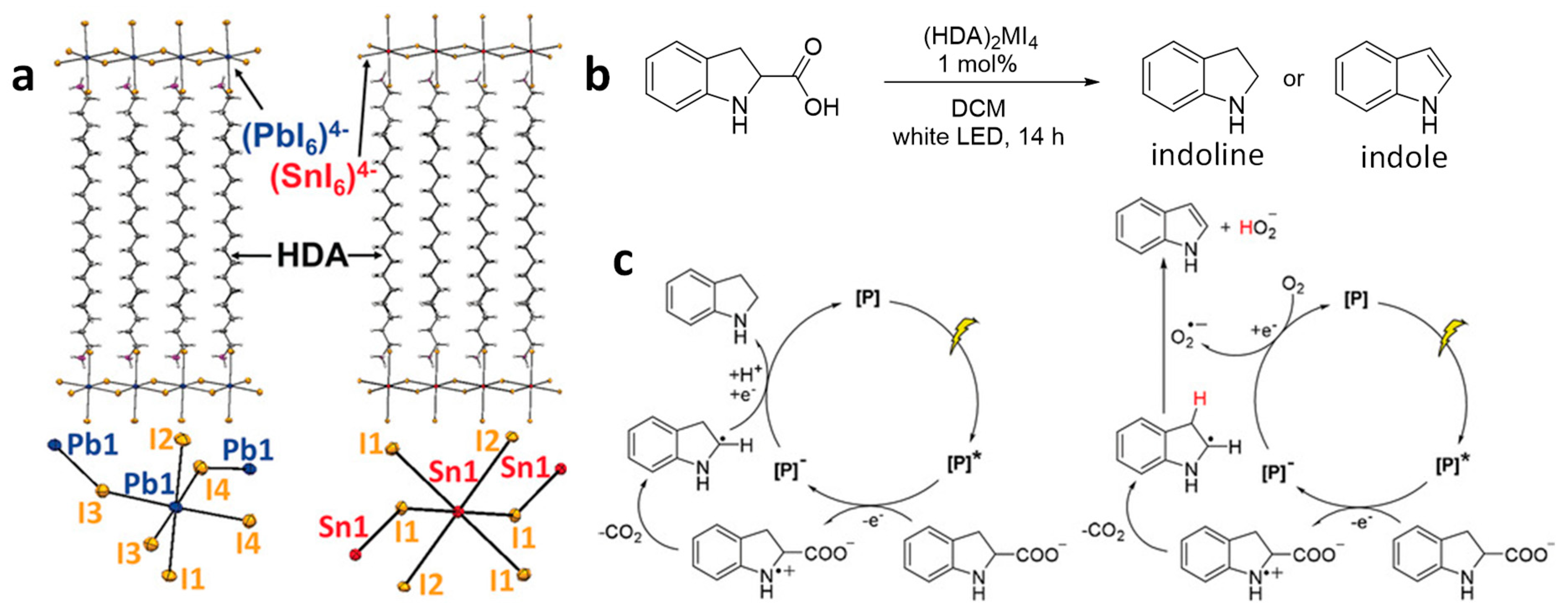
4.7. C–C Bond Cleavage via Decarboxylation and Dehydrogenation Reactions
5. Doped NCs
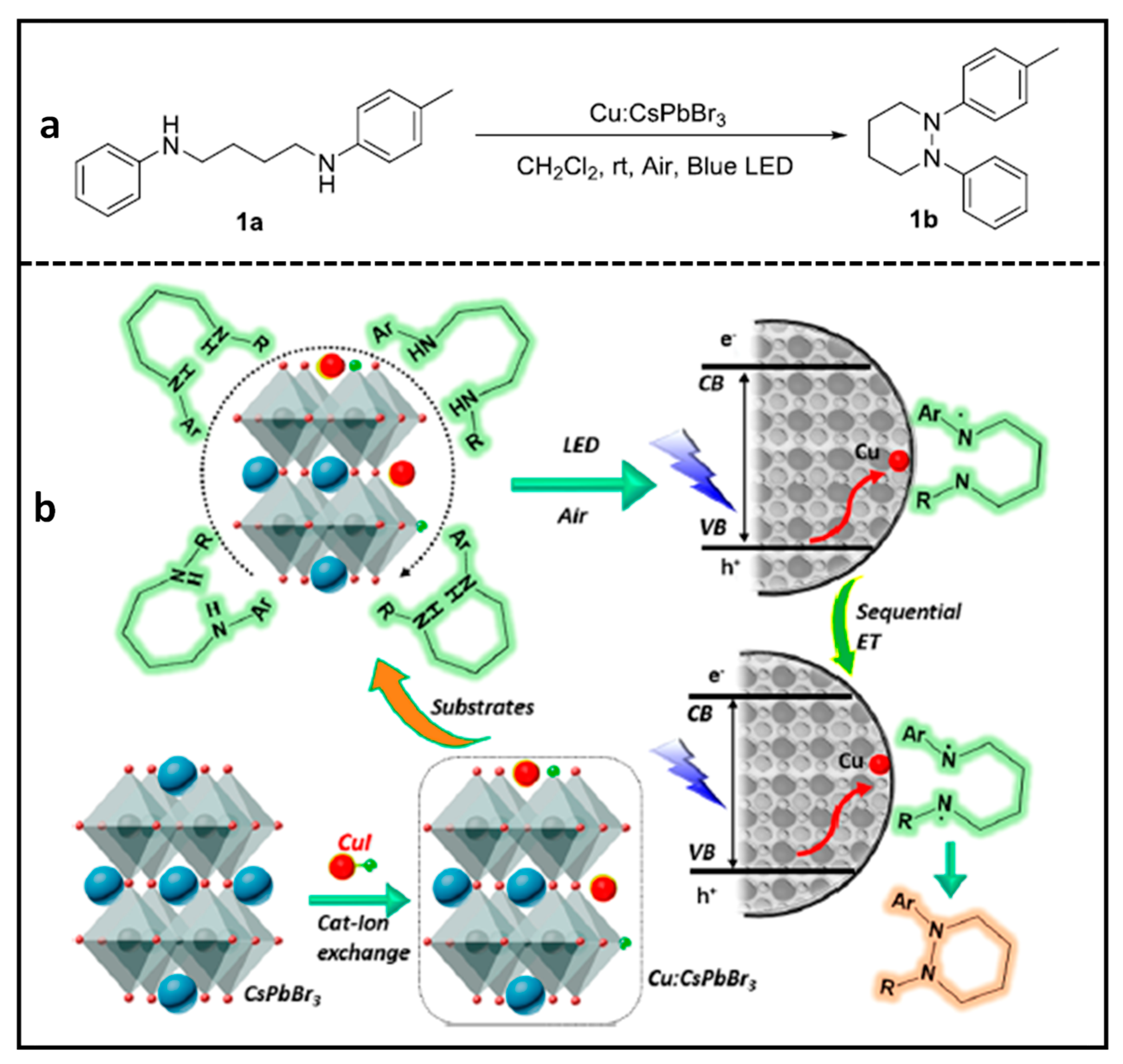
Cu-Doped CsPbBr3 NCs for N–N Heterocyclization Reaction
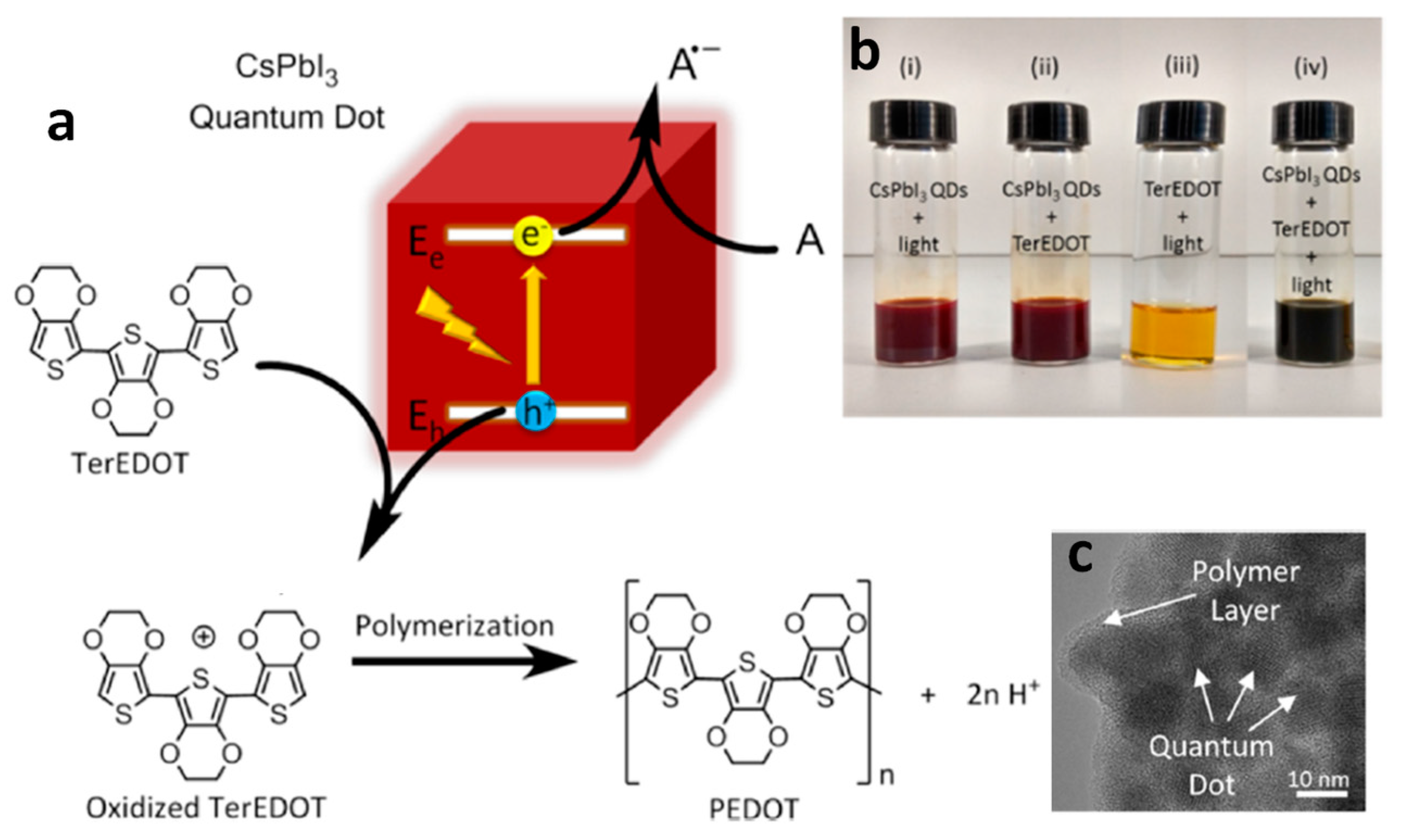
6. Photocatalytic Polymerization
7. Coupling of Thiols to Disulfides

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Irradiation | Organic Transformation/ Photocatalytic Reaction | C (%) | S (%) | Year | Ref |
|---|---|---|---|---|---|---|
| TiO2 | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 15 | 95 | 2018 | [92] |
| FAPbBr3 | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 15 | 99 | 2018 | [92] |
| nano-FAPbBr3 | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 11 | 99 | 2018 | [92] |
| 15% FAPbBr3/TiO2 | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 63 | 99 | 2018 | [92] |
| 15% FAPbBr3/SiO2 | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 13 | 99 | 2018 | [92] |
| 15% FAPbBr3/TiO2-M | simulated light irradiation, AM 1.5 G | oxidation of benzylic alcohol | 37 | 99 | 2018 | [92] |
| 15% FAPbBr3/TiO2 | λ ≥ 500 nm | oxidation of benzylic alcohol | 13 | 99 | 2018 | [92] |
| 15% FAPbBr3/TiO2 | without light irradiation | oxidation of benzylic alcohol | 0 | 0 | 2018 | [92] |
| CdS NCs | 2.6 mW 405 nm laser | oxidation of benzylic alcohol | - | 99 | 2019 | [93] |
| (Au@CdS)/Ni | 300 W Xe lamp (λ > 420 nm) | oxidation of benzylic alcohol | - | 99 | 2022 | [94] |
| Au2S/CdS | 6 W blue LED (445 nm) | oxidation of benzylic alcohol | 99 | 100 | 2022 | [95] |
| g-C3N4 | oxidation of benzylic alcohol | 99 | [96] | |||
| 5% NiOx/FAPbBr3/TiO2 | 150 W Xe lamp, AM 1.5 G simulated light irradiation | Oxidation of cyclohexane | 0.016 | >99 | 2019 | [97] |
| 5% NiOx/FAPbBr3/TiO2 | 150 W Xe lamp, AM 1.5 G simulated light irradiation | Oxidation of cyclooctane | 0.032 | >99 | 2019 | [97] |
| CsPbI3 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 58 | - | 2018 | [36] |
| CsPbBr3 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 98 | - | 2018 | [36] |
| CsPbBr2Cl | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 98 | - | 2018 | [36] |
| CsPbBr0.5Cl0.5 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 68 | - | 2018 | [36] |
| CsPbBrCl2 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 35 | - | 2018 | [36] |
| CsPbCl3 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 12 | 2018 | [36] | |
| CsPbCl3 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 93 | - | 2018 | [36] |
| CsPbCl3 + Br2 | Vis LED, 420–700 nm | thiophenol coupled to disulfide | 62 | - | 2018 | [36] |
| Cs3Bi2Br9 | visible light ≥ 420 nm | alcoholysis of styrene oxide in isopropanol (IPA) | >99 | - | 2019 | [98] |
| Cs3Bi2Br9 | visible light ≥ 420 nm | alcoholysis of styrene oxide in IPA | >99 | - | 2019 | [98] |
| CsPbBr3 | visible light ≥ 420 nm | alcoholysis of styrene oxide in IPA | 1 | - | 2019 | [98] |
| CsPbI3 | visible light ≥ 495 nm | polymerization of 3,4-ethylenedioxythiophene | 32.6 | - | 2017 | [86] |
| CsPbBr3 | Blue LED 455 nm | α-alkylation of aldehydes | >99 | 96 | 2019 | [65] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | synthesis aldehyde | 85 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | synthesis aldehyde | 52 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | synthesis tertiary amines | 90 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | synthesis tertiary amines | 79 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | cyclization of benzaldehyde phenylhydrazone | 88 | - | 2019 | [26] |
| MAPbBr3 | 12 W Blue LED, 455 nm | cyclization of benzaldehyde phenylhydrazone | 75 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | cyclization of ethyl (Z)-3-phenyl-3-(phenylamino)acrylate | 93 | - | 2019 | [26] |
| MAPbBr3 | 12 W Blue LED, 455 nm | cyclization of ethyl (Z)-3-phenyl-3-(phenylamino)acrylate | 65 | - | 2019 | [26] |
| CsPbBr3 NCs | 12 W Blue LED, 455 nm | coupling of benzoic acid with 4-bromotrifluorobenzene | 78 | - | 2019 | [26] |
| CsPbBr3 | 4.6 W Blue LED | photopolymerized styrene | 12 | - | 2018 | [99] |
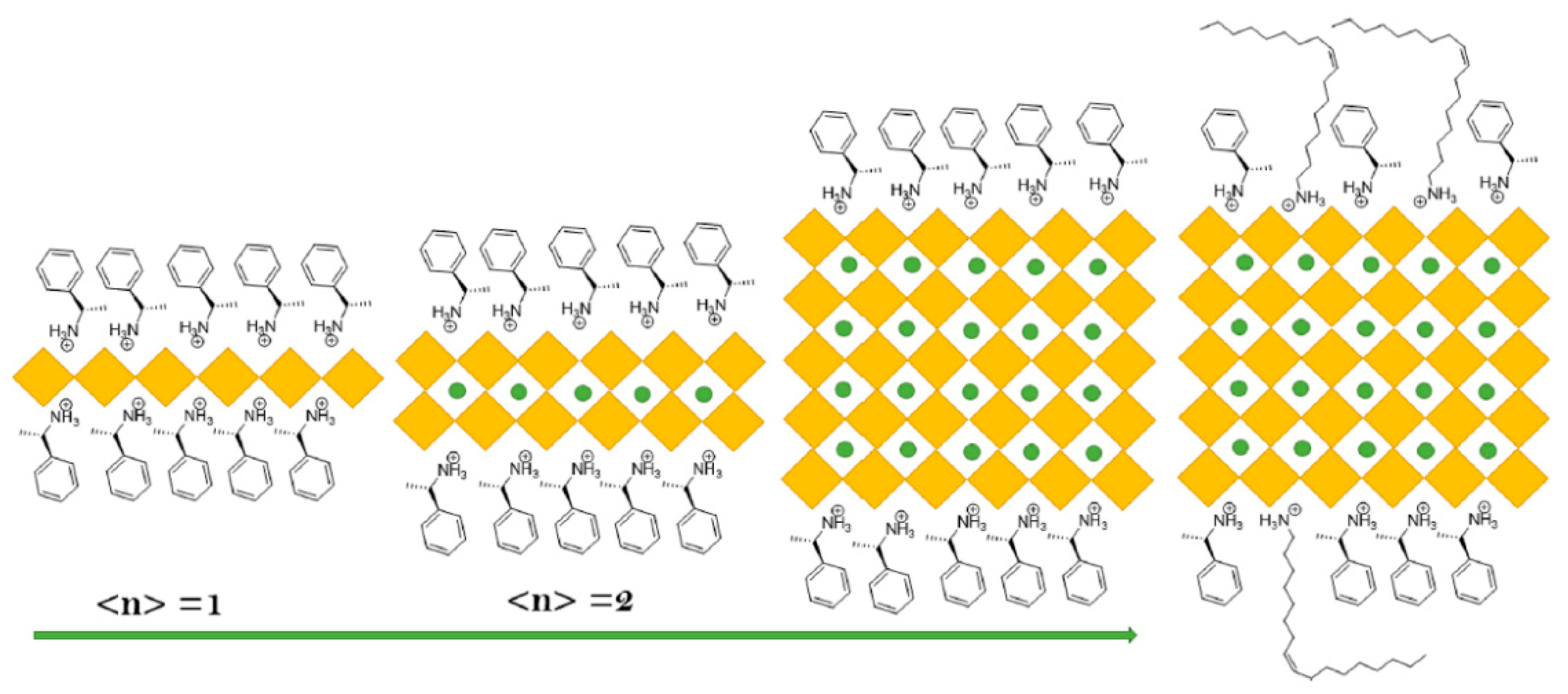
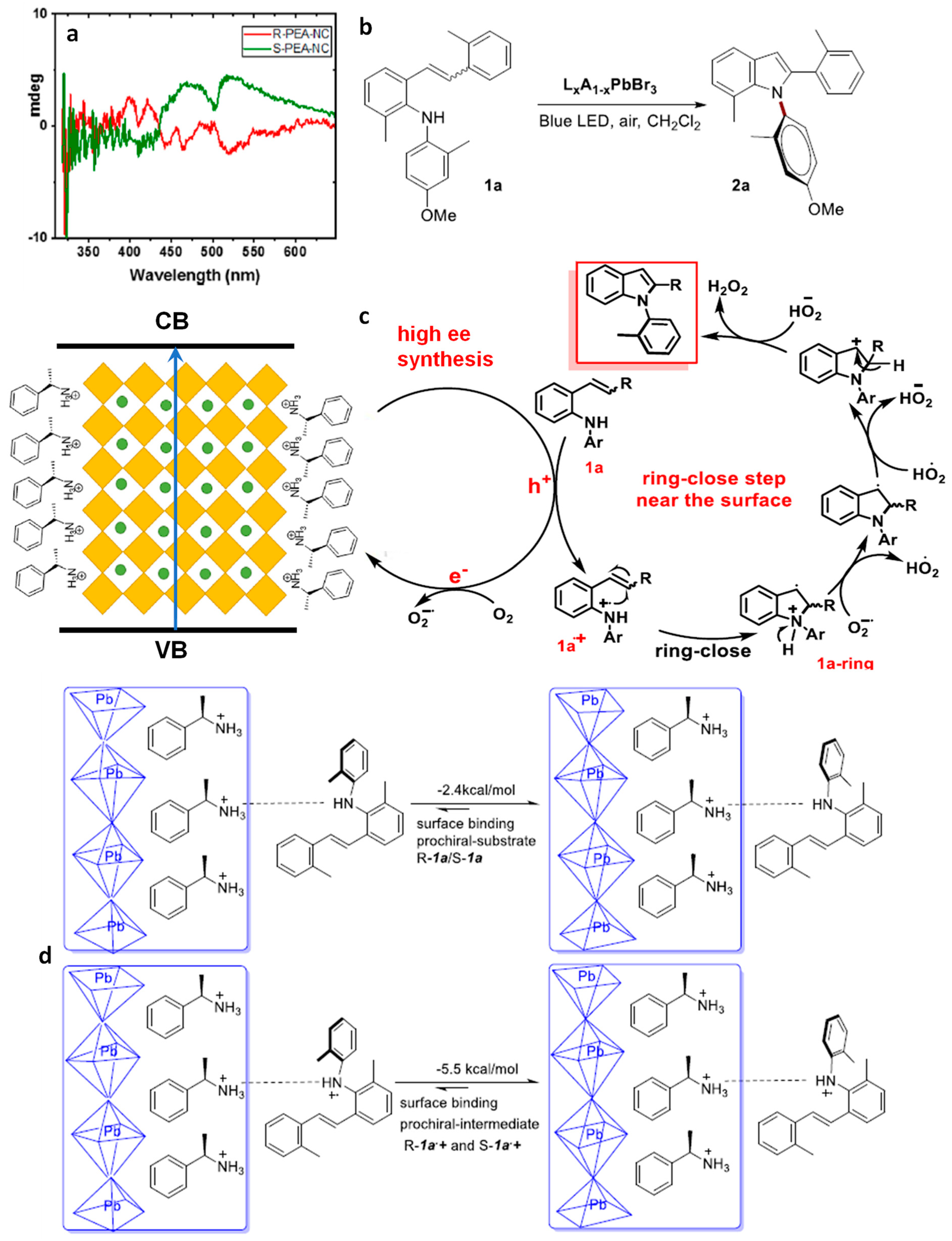
8. Chiral Perovskite NCs
Production of N–C Axial Chiral N-Heterocycles Using Chiral CsPbBr3 NCs
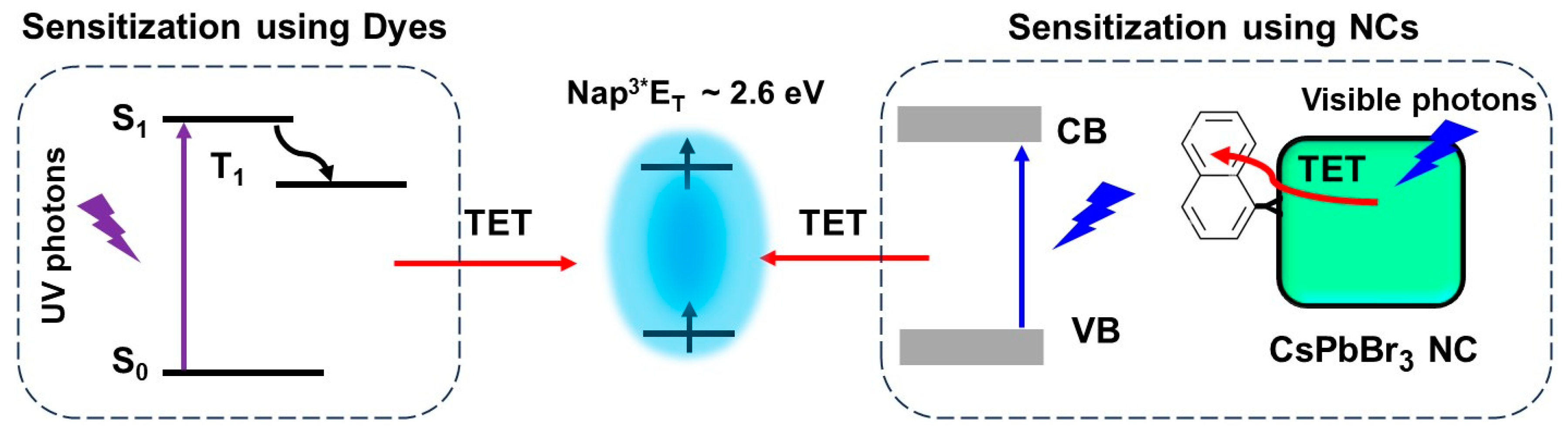
9. Triplet Energy Transfer from Dyes and NCs to Substrates
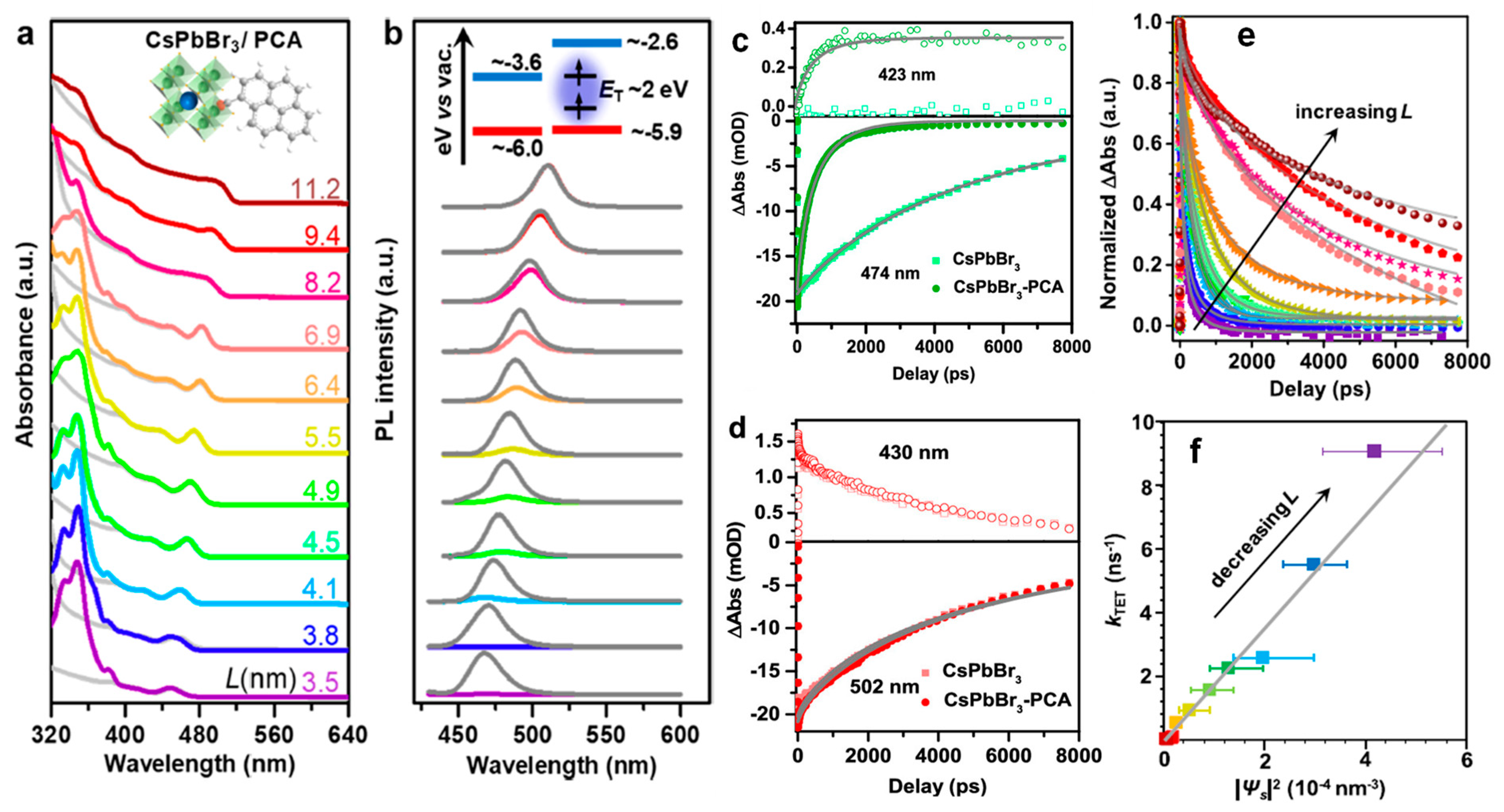
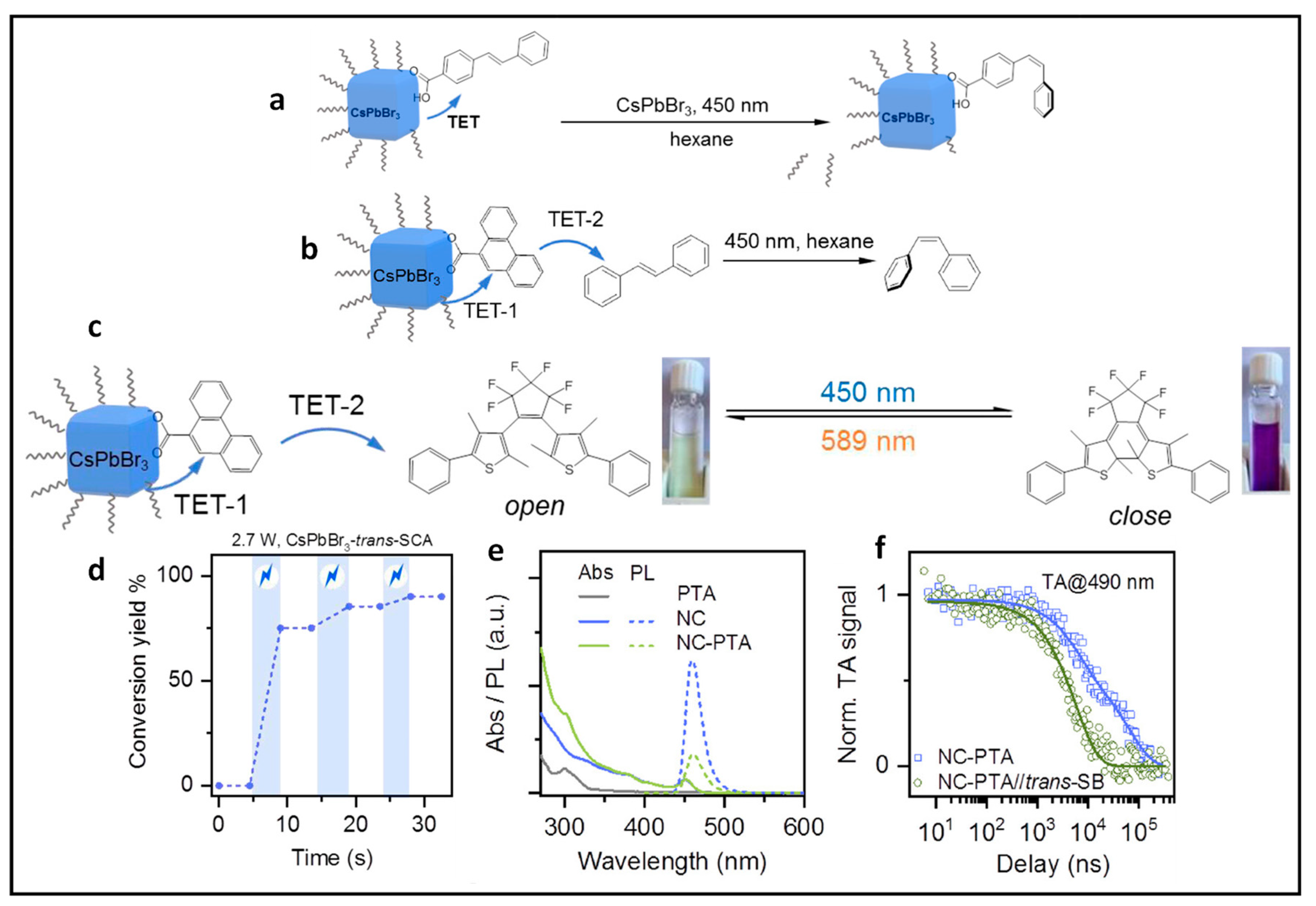
9.1. Demonstration of TET from Quantum Confined PNCs to Substrates
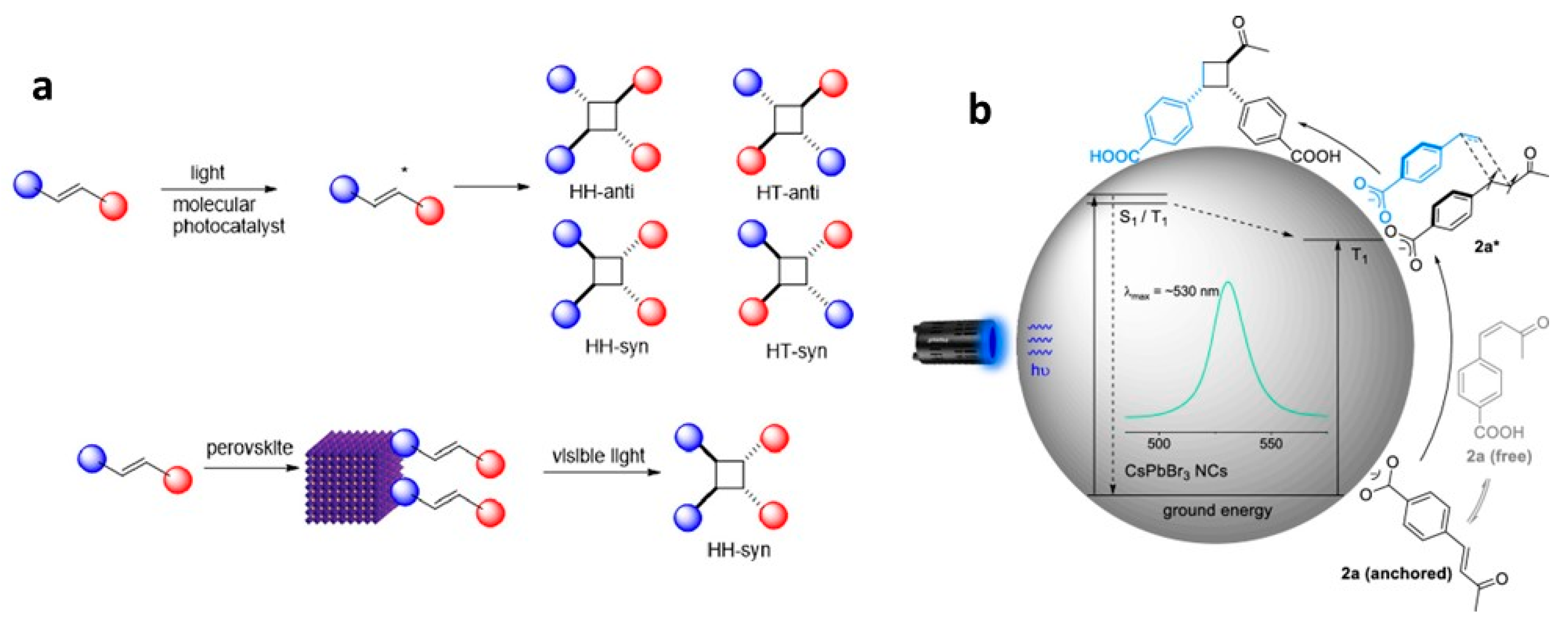
9.2. Isomerisation of Surface-Anchored Stilbene
9.3. Isomerization of Stilbene in Solution Using Relay
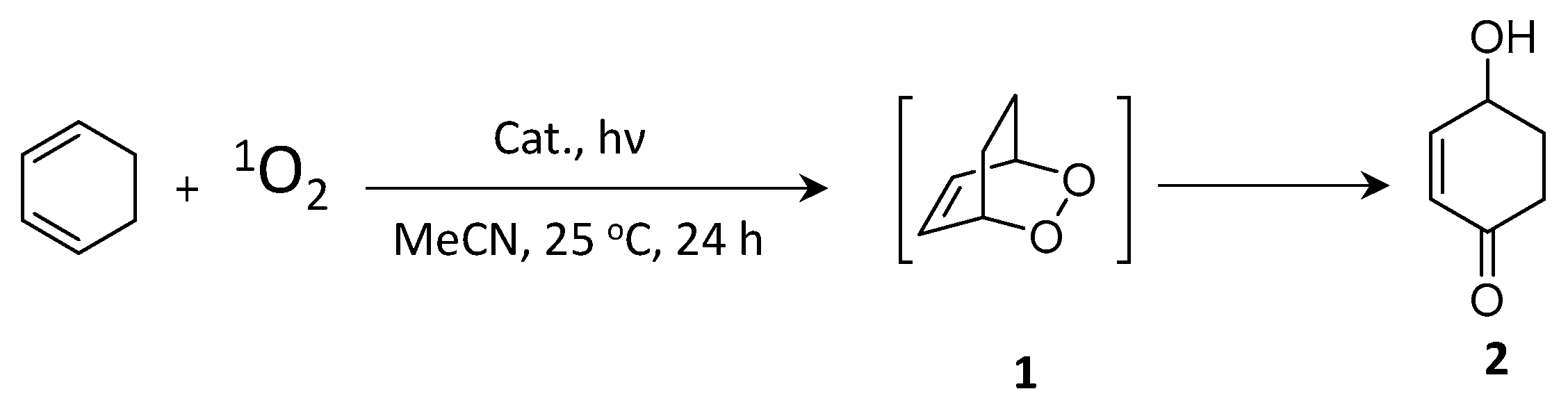
9.4. Photocatalytic 2 + 2 Cycloadditions
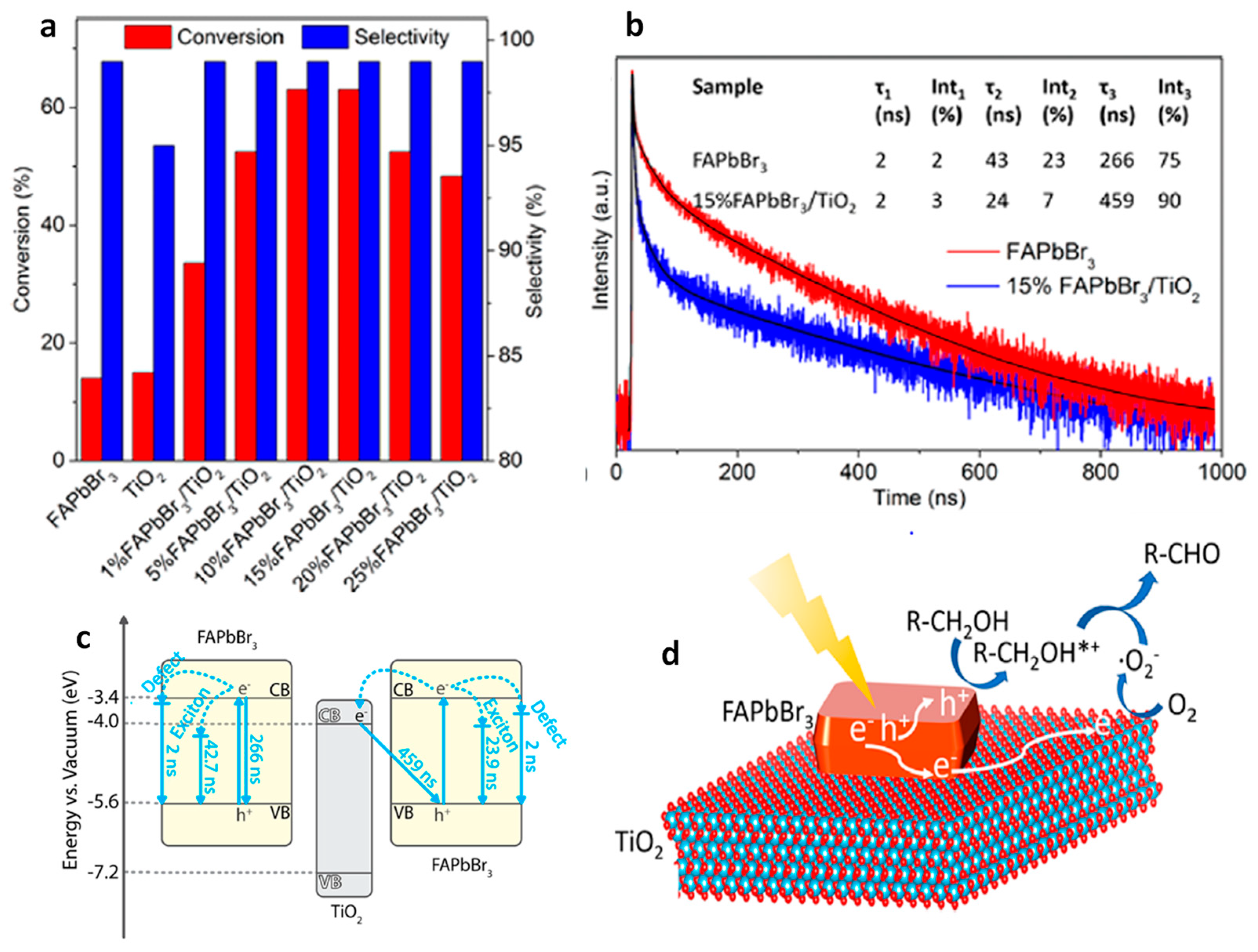
10. Benzaldehyde Formation from Oxidation of Benzyl Alcohol and Toluene
Benzaldehyde Formation Using Pb-Based Perovskite Heterostructures as Photocatalyst
11. Preparation of Singlet Oxygen Driven Oxidized Compounds Using g-C3N4/Perovskite
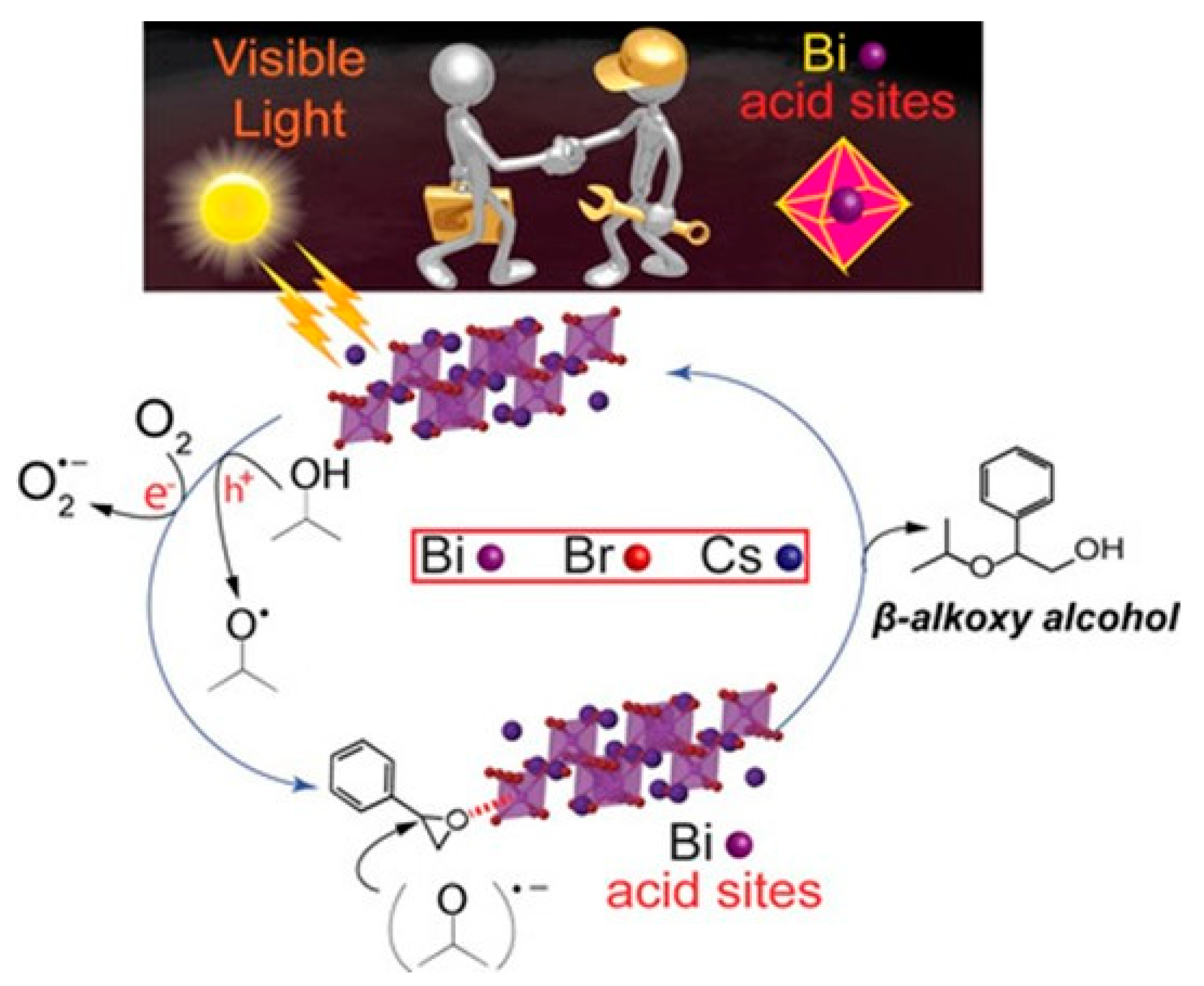
12. Opening of Epoxide Reactions Using Lead-Free Cs3Bi2Br9 Perovskites
13. Benzaldehyde Formation by Toluene Oxidation
13.1. Lead-Free Cs3Bi2Br9 Perovskites
13.2. Cs3Bi2Br9/Pd Heterostructures
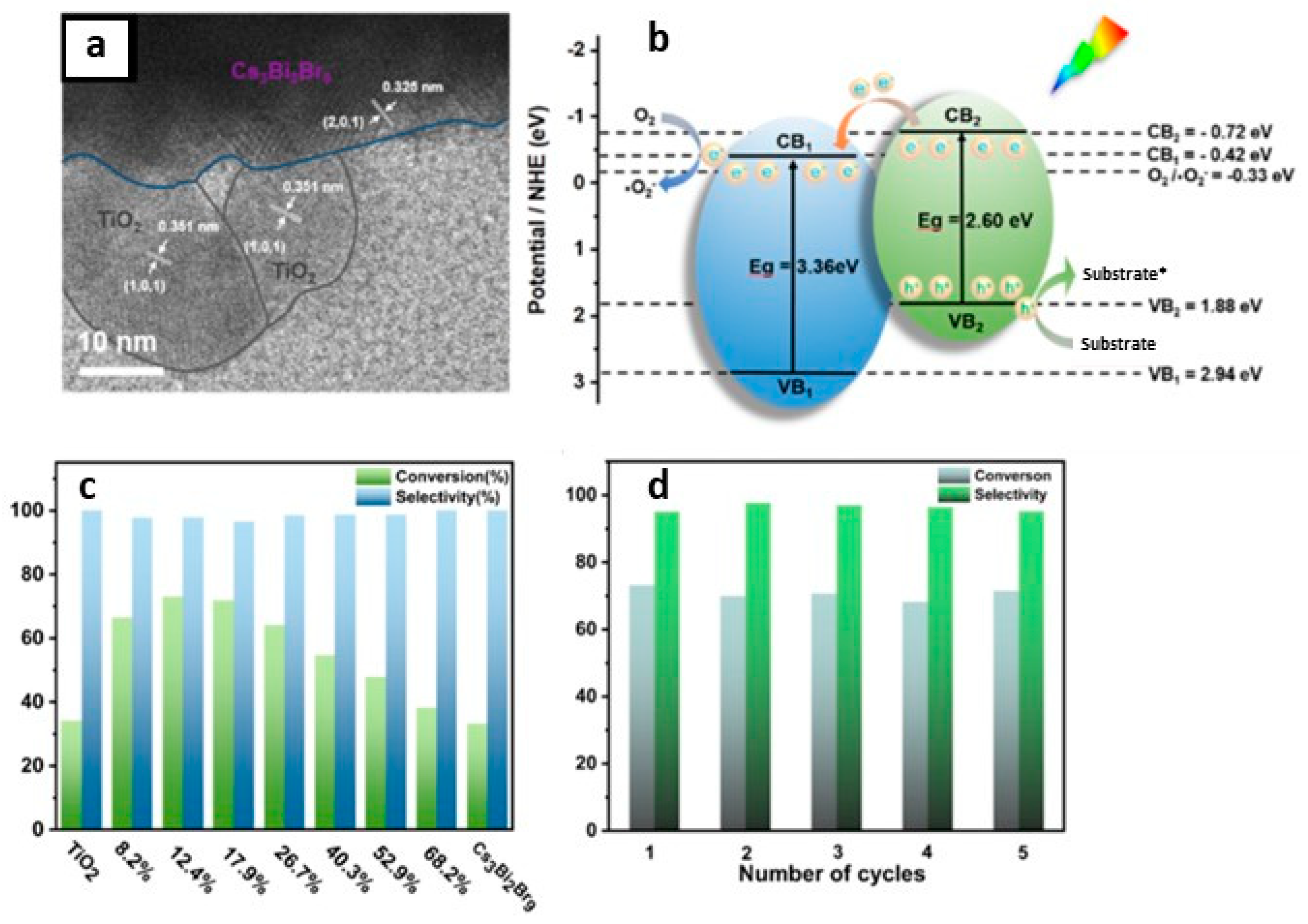
13.3. Cs3Bi2Br9/TiO2 Heterostructures
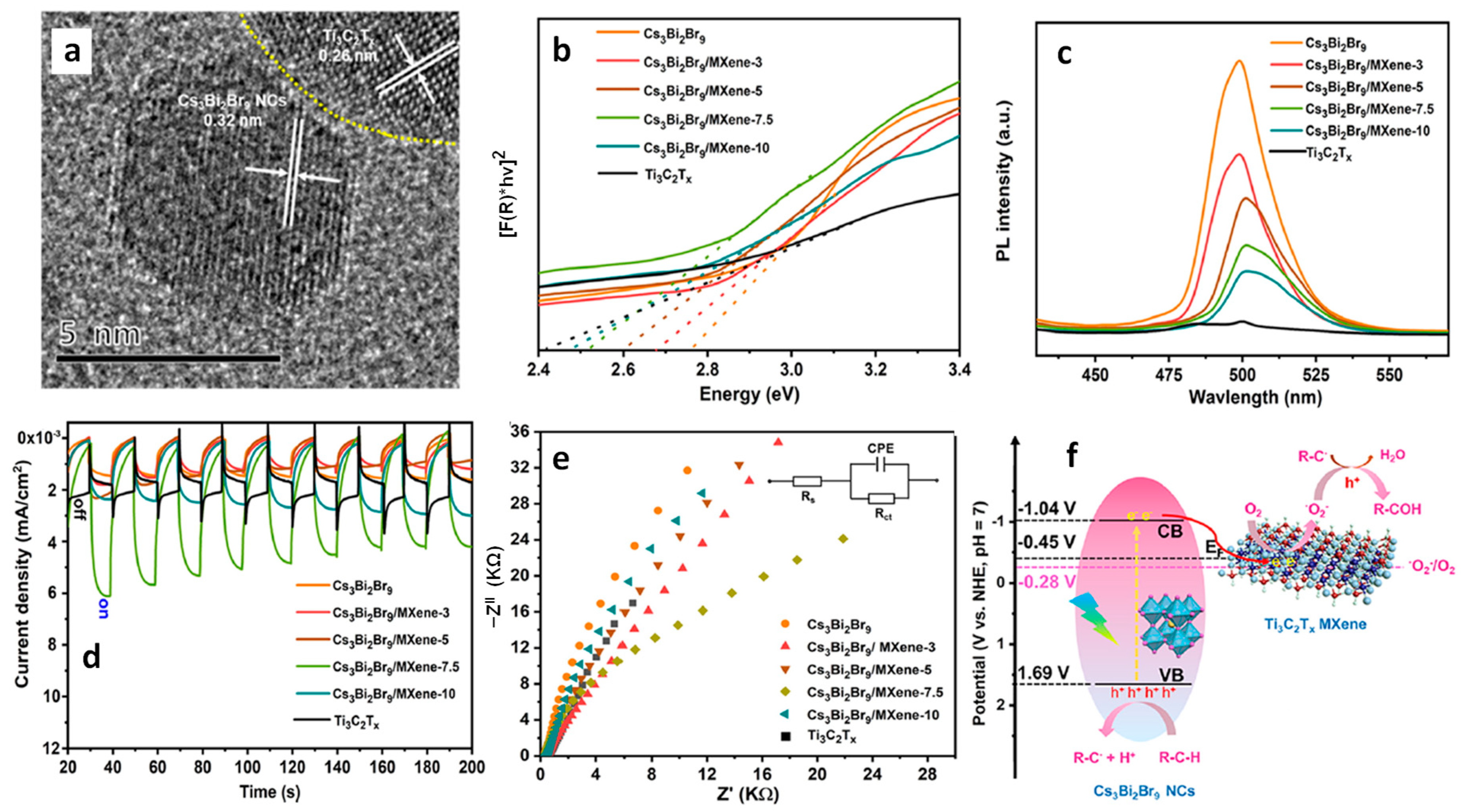
13.4. Cs3Bi2Br9/MXene Heterostructures
13.5. Cs3Bi2Br9/g-C3N4 Heterostructures
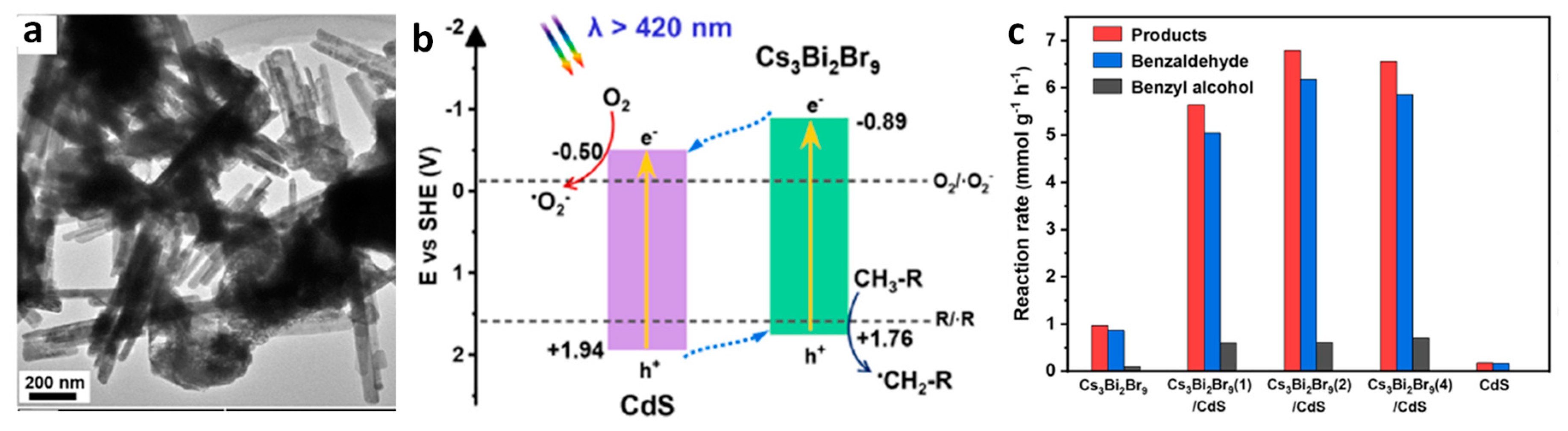
13.6. Cs3Bi2Br9/CdS Heterostructures
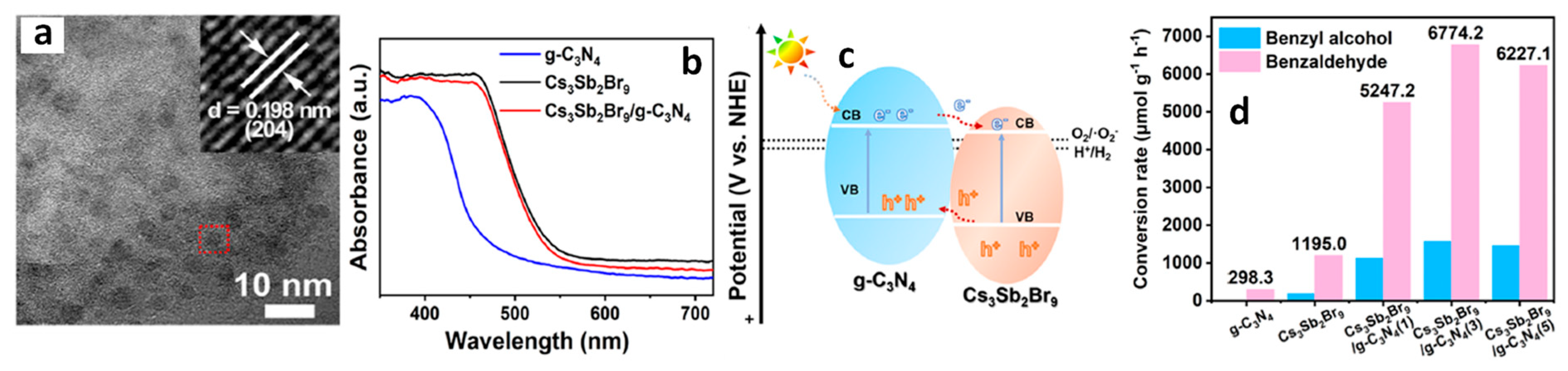
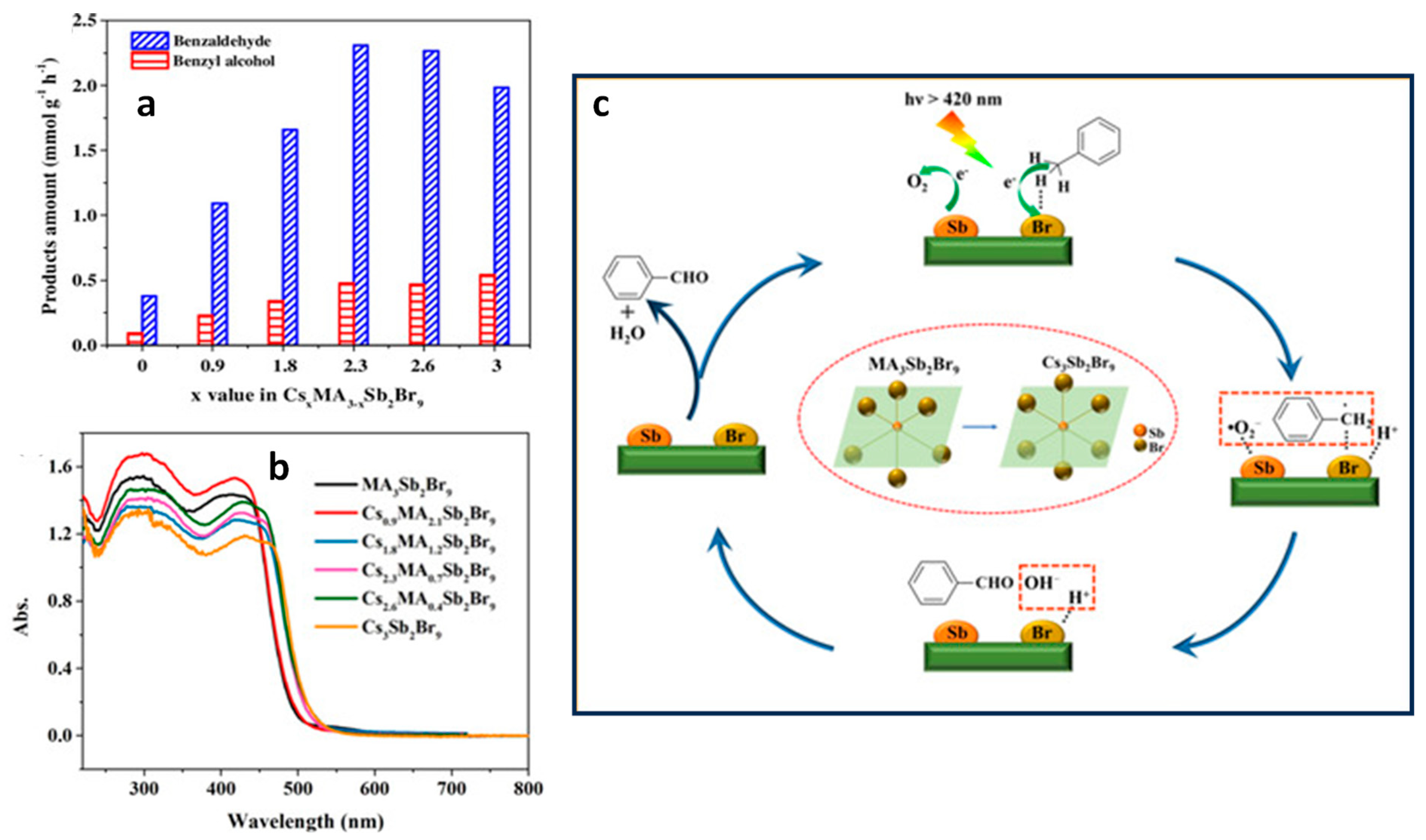
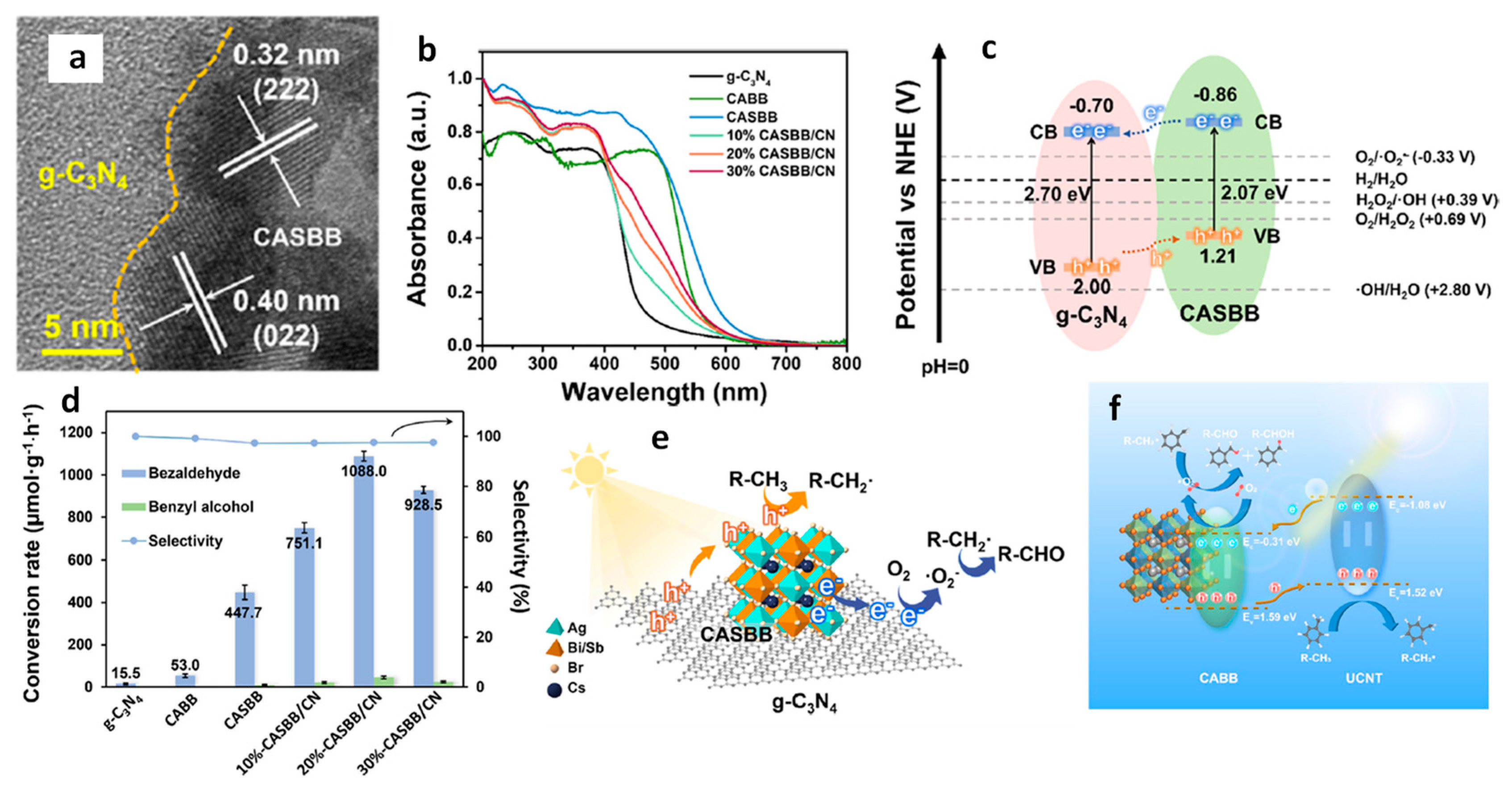
13.7. Cs3Sb2Br9/g-C3N4 Heterostructures
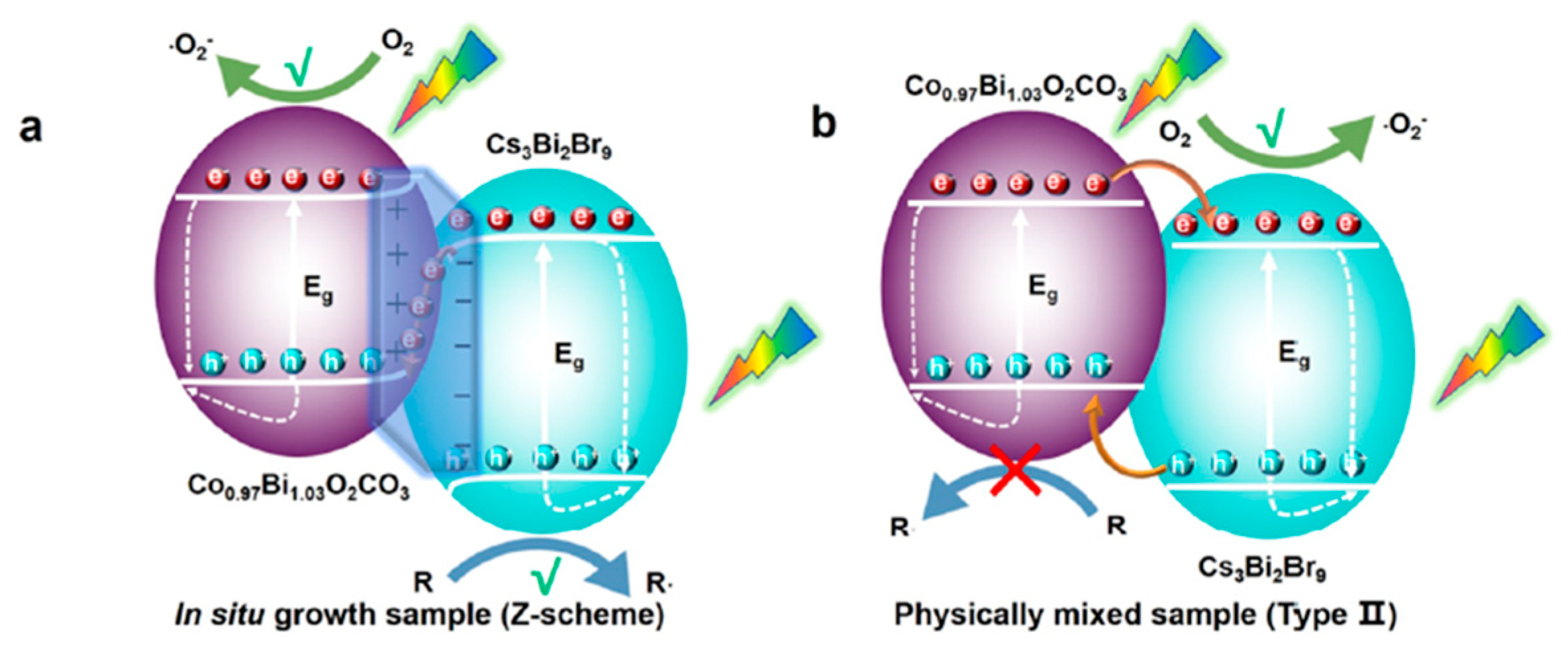
13.8. Z-Scheme Heterojunction Formation by Cosharing Atoms
13.9. Z-Scheme Heterojunction of Cs3Bi2Br9/d-BiOBr
13.10. Sb-Doped Cs3Bi2Br9 Perovskite
13.11. Effect of A-Site Cation in A3Sb2Br9
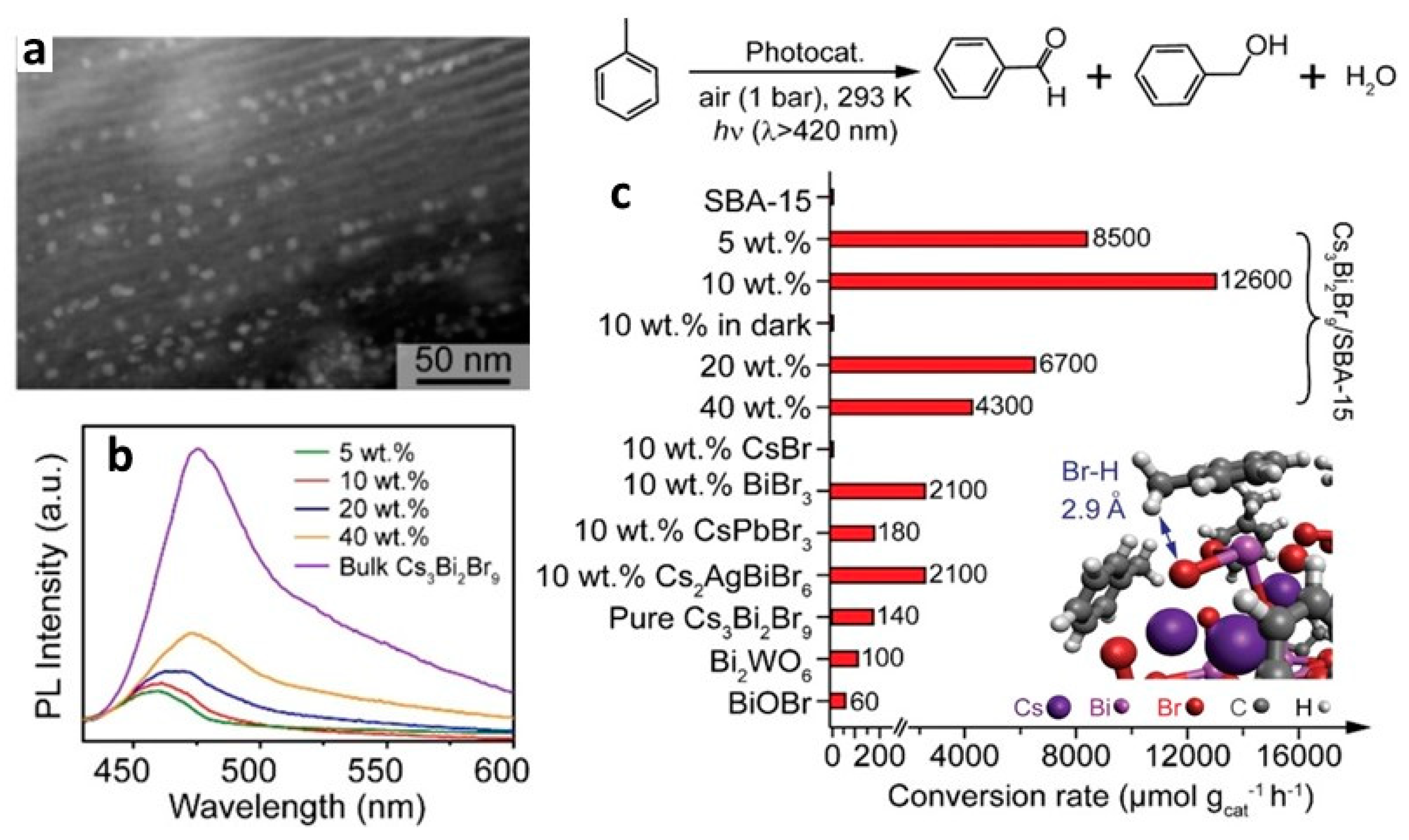
13.12. Cs3Bi2Br9/SiO2 Heterostructures
13.13. DP NCs
13.14. A4MIIM2IIIX12 LDP NCs
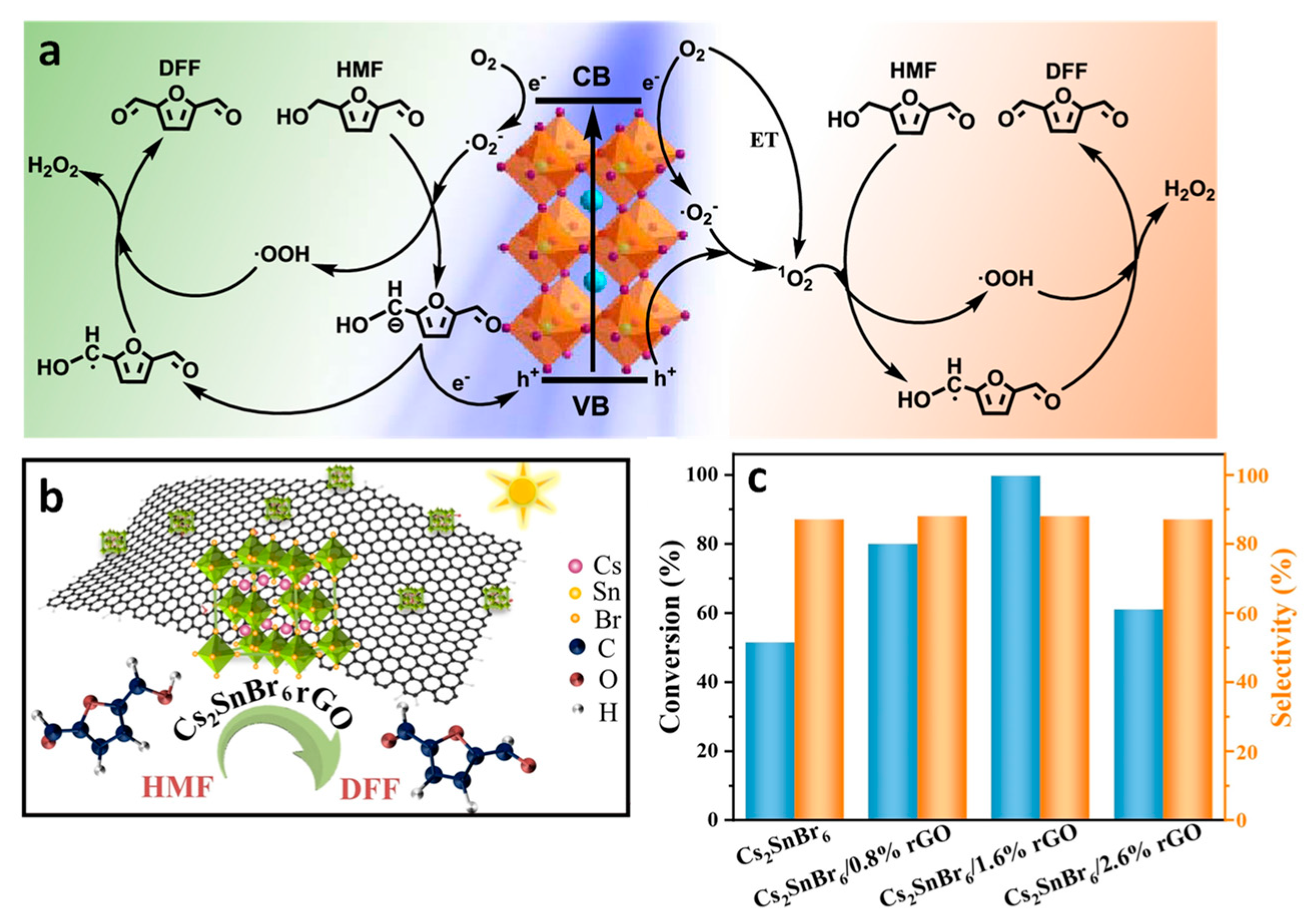
14. Oxidation of 5-Hydroxymethylfurfural to 2,5-Diformylfuran
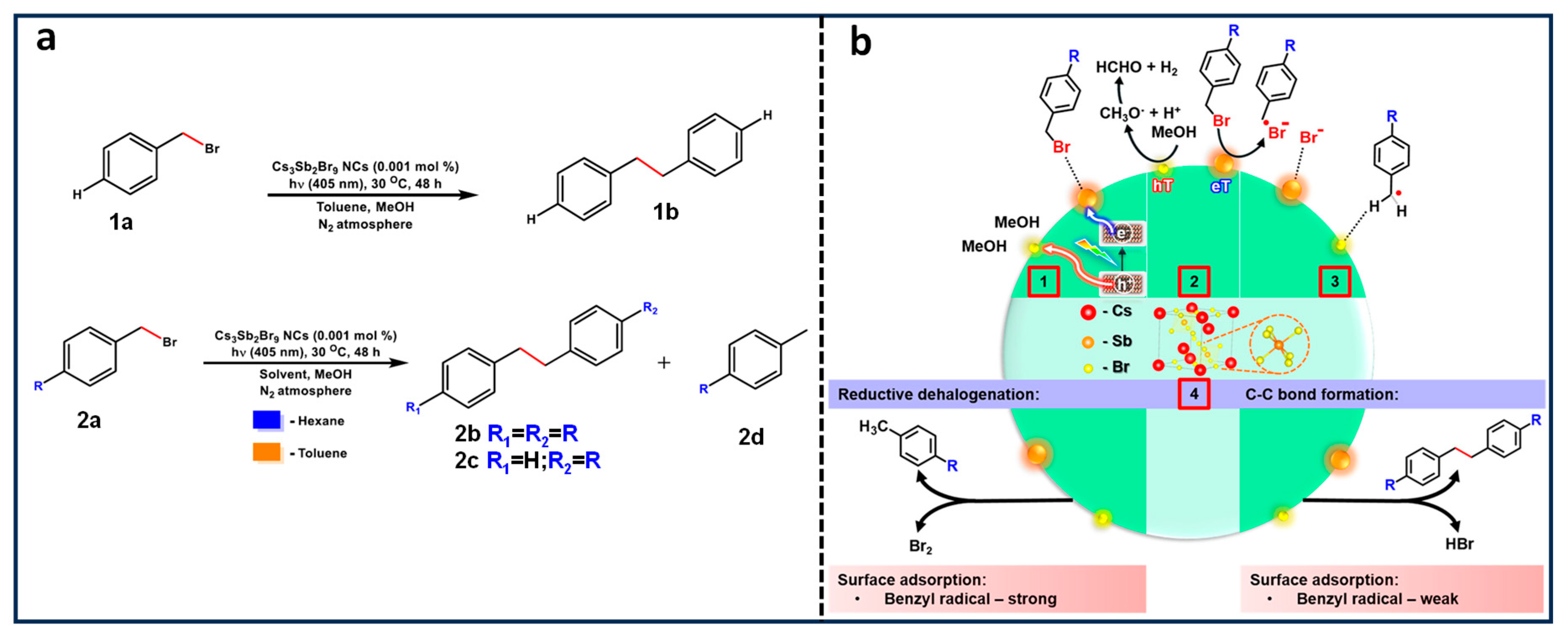
15. Csp3-Csp3 Coupling Reaction Using Lead-Free Cs3Sb2Br9 NCs
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, W.; Tadé, M.O.; Shao, Z. Research progress of perovskite materials in photocatalysis- and photovoltaics-related energy conversion and environmental treatment. Chem. Soc. Rev. 2015, 44, 5371–5408. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Xiao, W.-J. Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Gisbertz, S.; Pieber, B. Heterogeneous Photocatalysis in Organic Synthesis. ChemPhotoChem 2020, 4, 456–475. [Google Scholar] [CrossRef]
- Caputo, J.A.; Frenette, L.C.; Zhao, N.; Sowers, K.L.; Krauss, T.D.; Weix, D.J. General and Efficient C–C Bond Forming Photoredox Catalysis with Semiconductor Quantum Dots. J. Am. Chem. Soc. 2017, 139, 4250–4253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-B.; Li, Z.-J.; Gao, Y.-J.; Meng, Q.-Y.; Yu, S.; Weiss, R.G.; Tung, C.-H.; Wu, L.-Z. Mechanistic Insights into the Interface-Directed Transformation of Thiols into Disulfides and Molecular Hydrogen by Visible-Light Irradiation of Quantum Dots. Angew. Chem. Int. Ed. 2014, 53, 2085–2089. [Google Scholar] [CrossRef]
- Chai, Z.; Zeng, T.-T.; Li, Q.; Lu, L.-Q.; Xiao, W.-J.; Xu, D. Efficient Visible Light-Driven Splitting of Alcohols into Hydrogen and Corresponding Carbonyl Compounds over a Ni-Modified CdS Photocatalyst. J. Am. Chem. Soc. 2016, 138, 10128–10131. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, H.; Wang, S.; Li, X. How to apply metal halide perovskites to photocatalysis: Challenges and development. Nanoscale 2021, 13, 10281–10304. [Google Scholar] [CrossRef]
- Jin, N.; Sun, Y.; Shi, W.; Wang, P.; Nagaoka, Y.; Cai, T.; Wu, R.; Dube, L.; Nyiera, H.N.; Liu, Y.; et al. Type-I CdS/ZnS Core/Shell Quantum Dot-Gold Heterostructural Nanocrystals for Enhanced Photocatalytic Hydrogen Generation. J. Am. Chem. Soc. 2023, 145, 21886–21896. [Google Scholar] [CrossRef]
- Cai, T.; Shi, W.; Hwang, S.; Kobbekaduwa, K.; Nagaoka, Y.; Yang, H.; Hills-Kimball, K.; Zhu, H.; Wang, J.; Wang, Z.; et al. Lead-Free Cs4CuSb2Cl12 Layered Double Perovskite Nanocrystals. J. Am. Chem. Soc. 2020, 142, 11927–11936. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bodnarchuk, M.I.; Kershaw, S.V.; Kovalenko, M.V.; Rogach, A.L. Lead Halide Perovskite Nanocrystals in the Research Spotlight: Stability and Defect Tolerance. ACS Energy Lett. 2017, 2, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.K.; Markad, G.B.; Nag, A. Band Edge Energies and Excitonic Transition Probabilities of Colloidal CsPbX3 (X = Cl, Br, I) Perovskite Nanocrystals. ACS Energy Lett. 2016, 1, 665–671. [Google Scholar] [CrossRef]
- Xing, G.; Mathews, N.; Sun, S.; Lim, S.S.; Lam, Y.M.; Grätzel, M.; Mhaisalkar, S.; Sum, T.C. Long-Range Balanced Electron- and Hole-Transport Lengths in Organic-Inorganic CH3NH3PbI3. Science 2013, 342, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Yettapu, G.R.; Talukdar, D.; Sarkar, S.; Swarnkar, A.; Nag, A.; Ghosh, P.; Mandal, P. Terahertz Conductivity within Colloidal CsPbBr3 Perovskite Nanocrystals: Remarkably High Carrier Mobilities and Large Diffusion Lengths. Nano Lett. 2016, 16, 4838–4848. [Google Scholar] [CrossRef] [PubMed]
- Stranks, S.D.; Eperon, G.E.; Grancini, G.; Menelaou, C.; Alcocer, M.J.P.; Leijtens, T.; Herz, L.M.; Petrozza, A.; Snaith, H.J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Subramanian Periyal, S.; Jagadeeswararao, M.; Ng, S.E.; John, R.A.; Mathews, N. Halide Perovskite Quantum Dots Photosensitized-Amorphous Oxide Transistors for Multimodal Synapses. Adv. Mater. Technol. 2020, 5, 2000514. [Google Scholar] [CrossRef]
- Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 19 November 2023).
- Filip, M.R.; Verdi, C.; Giustino, F. GW Band Structures and Carrier Effective Masses of CH3NH3PbI3 and Hypothetical Perovskites of the Type APbI3: A = NH4, PH4, AsH4, and SbH4. J. Phys. Chem. C 2015, 119, 25209–25219. [Google Scholar] [CrossRef]
- Vashishtha, P.; Bishnoi, S.; Li, C.H.A.; Jagadeeswararao, M.; Hooper, T.J.N.; Lohia, N.; Shivarudraiah, S.B.; Ansari, M.S.; Sharma, S.N.; Halpert, J.E. Recent Advancements in Near-Infrared Perovskite Light-Emitting Diodes. ACS Appl. Electron. Mater. 2020, 2, 3470–3490. [Google Scholar] [CrossRef]
- John, R.A.; Shah, N.; Vishwanath, S.K.; Ng, S.E.; Febriansyah, B.; Jagadeeswararao, M.; Chang, C.-H.; Basu, A.; Mathews, N. Halide perovskite memristors as flexible and reconfigurable physical unclonable functions. Nat. Commun. 2021, 12, 3681. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chang, W.J.; Lee, C.W.; Park, S.; Ahn, H.-Y.; Nam, K.T. Photocatalytic hydrogen generation from hydriodic acid using methylammonium lead iodide in dynamic equilibrium with aqueous solution. Nat. Energy 2016, 2, 16185. [Google Scholar] [CrossRef]
- Temerov, F.; Baghdadi, Y.; Rattner, E.; Eslava, S. A Review on Halide Perovskite-Based Photocatalysts: Key Factors and Challenges. ACS Appl. Energy Mater. 2022, 5, 14605–14637. [Google Scholar] [CrossRef] [PubMed]
- Ketavath, R.; Mohan, L.; Sumukam, R.R.; Alsulami, Q.A.; Premalatha, A.; Murali, B. Can perovskites be efficient photocatalysts in organic transformations? J. Mater. Chem. A 2022, 10, 12317–12333. [Google Scholar] [CrossRef]
- Zhu, X.; Lin, Y.; San Martin, J.; Sun, Y.; Zhu, D.; Yan, Y. Lead halide perovskites for photocatalytic organic synthesis. Nat. Commun. 2019, 10, 2843. [Google Scholar] [CrossRef] [PubMed]
- Vargas, B.; Ramos, E.; Pérez-Gutiérrez, E.; Alonso, J.C.; Solis-Ibarra, D. A Direct Bandgap Copper–Antimony Halide Perovskite. J. Am. Chem. Soc. 2017, 139, 9116–9119. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Chakraborty, R.; Ghosh, P.; Nag, A. Low-Bandgap Cs4CuSb2Cl12 Layered Double Perovskite: Synthesis, Reversible Thermal Changes, and Magnetic Interaction. Chem.–Asian J. 2018, 13, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Mai, H.; Li, X.; Lu, J.; Wen, X.; Le, T.C.; Russo, S.P.; Chen, D.; Caruso, R.A. Synthesis of Layered Lead-Free Perovskite Nanocrystals with Precise Size and Shape Control and Their Photocatalytic Activity. J. Am. Chem. Soc. 2023, 145, 17337–17350. [Google Scholar] [CrossRef]
- Huang, H.; Pradhan, B.; Hofkens, J.; Roeffaers, M.B.J.; Steele, J.A. Solar-Driven Metal Halide Perovskite Photocatalysis: Design, Stability, and Performance. ACS Energy Lett. 2020, 5, 1107–1123. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, R.; Li, D.; Jiang, Y.; Wang, X.; Tang, H.; Xu, J. Recent Progress in Halide Perovskite Nanocrystals for Photocatalytic Hydrogen Evolution. Nanomaterials 2023, 13, 106. [Google Scholar] [CrossRef]
- Dandia, A.; Saini, P.; Sharma, R.; Parewa, V. Visible light driven perovskite-based photocatalysts: A new candidate for green organic synthesis by photochemical protocol. Curr. Res. Green Sustain. Chem. 2020, 3, 100031. [Google Scholar] [CrossRef]
- Bresolin, B.-M.; Park, Y.; Bahnemann, D.W. Recent Progresses on Metal Halide Perovskite-Based Material as Potential Photocatalyst. Catalysts 2020, 10, 709. [Google Scholar] [CrossRef]
- Ren, K.; Yue, S.; Li, C.; Fang, Z.; Gasem, K.A.M.; Leszczynski, J.; Qu, S.; Wang, Z.; Fan, M. Metal halide perovskites for photocatalysis applications. J. Mater. Chem. A 2022, 10, 407–429. [Google Scholar] [CrossRef]
- Wang, X.; Peng, Y.; Yang, S.; Yang, H.G.; Hou, Y. Recent progress in metal halide perovskite photocatalysts for hydrogen evolution. Mater. Chem. Front. 2023, 7, 4635–4657. [Google Scholar] [CrossRef]
- Wu, W.-B.; Wong, Y.-C.; Tan, Z.-K.; Wu, J. Photo-induced thiol coupling and C–H activation using nanocrystalline lead-halide perovskite catalysts. Catal. Sci. Technol. 2018, 8, 4257–4263. [Google Scholar] [CrossRef]
- Protesescu, L.; Yakunin, S.; Bodnarchuk, M.I.; Krieg, F.; Caputo, R.; Hendon, C.H.; Yang, R.X.; Walsh, A.; Kovalenko, M.V. Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut. Nano Lett. 2015, 15, 3692–3696. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Ye, J.; De, A.; Debroye, E.; Ha, S.K.; Bladt, E.; Kshirsagar, A.S.; Wang, Z.; Yin, J.; Wang, Y.; et al. State of the Art and Prospects for Halide Perovskite Nanocrystals. ACS Nano 2021, 15, 10775–10981. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.C.; Pertegás, A.; González-Carrero, S.; Malinkiewicz, O.; Agouram, S.; Mínguez Espallargas, G.; Bolink, H.J.; Galian, R.E.; Pérez-Prieto, J. Nontemplate Synthesis of CH3NH3PbBr3 Perovskite Nanoparticles. J. Am. Chem. Soc. 2014, 136, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Butkus, J.; Vashishtha, P.; Chen, K.; Gallaher, J.K.; Prasad, S.K.K.; Metin, D.Z.; Laufersky, G.; Gaston, N.; Halpert, J.E.; Hodgkiss, J.M. The Evolution of Quantum Confinement in CsPbBr3 Perovskite Nanocrystals. Chem. Mater. 2017, 29, 3644–3652. [Google Scholar] [CrossRef]
- Yang, B.; Chen, J.; Yang, S.; Hong, F.; Sun, L.; Han, P.; Pullerits, T.; Deng, W.; Han, K. Lead-Free Silver-Bismuth Halide Double Perovskite Nanocrystals. Angew. Chem. Int. Ed. 2018, 57, 5359–5363. [Google Scholar] [CrossRef]
- Huang, S.; Li, Z.; Kong, L.; Zhu, N.; Shan, A.; Li, L. Enhancing the Stability of CH3NH3PbBr3 Quantum Dots by Embedding in Silica Spheres Derived from Tetramethyl Orthosilicate in “Waterless” Toluene. J. Am. Chem. Soc. 2016, 138, 5749–5752. [Google Scholar] [CrossRef]
- Zhong, Q.; Cao, M.; Hu, H.; Yang, D.; Chen, M.; Li, P.; Wu, L.; Zhang, Q. One-Pot Synthesis of Highly Stable CsPbBr3@SiO2 Core–Shell Nanoparticles. ACS Nano 2018, 12, 8579–8587. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lan, X.; Ren, C.; Qi, B.; Shi, J. Multi-dimensional collaborations boost lead halide perovskite driven superior and long-period CO2 photoreduction under liquid-phase H2O environment. J. Alloys Compd. 2022, 899, 163316. [Google Scholar] [CrossRef]
- van der Stam, W.; Geuchies, J.J.; Altantzis, T.; van den Bos, K.H.W.; Meeldijk, J.D.; Van Aert, S.; Bals, S.; Vanmaekelbergh, D.; de Mello Donega, C. Highly Emissive Divalent-Ion-Doped Colloidal CsPb1–xMxBr3 Perovskite Nanocrystals through Cation Exchange. J. Am. Chem. Soc. 2017, 139, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, Z.; Tan, Z.; Song, H.; Ge, C.; Niu, G.; Han, J.; Tang, J. Rare Earth Ion-Doped CsPbBr3 Nanocrystals. Adv. Opt. Mater. 2018, 6, 1700864. [Google Scholar] [CrossRef]
- Pan, G.; Bai, X.; Yang, D.; Chen, X.; Jing, P.; Qu, S.; Zhang, L.; Zhou, D.; Zhu, J.; Xu, W.; et al. Doping Lanthanide into Perovskite Nanocrystals: Highly Improved and Expanded Optical Properties. Nano Lett. 2017, 17, 8005–8011. [Google Scholar] [CrossRef] [PubMed]
- Mir, W.J.; Mahor, Y.; Lohar, A.; Jagadeeswararao, M.; Das, S.; Mahamuni, S.; Nag, A. Postsynthesis Doping of Mn and Yb into CsPbX3 (X = Cl, Br, or I) Perovskite Nanocrystals for Downconversion Emission. Chem. Mater. 2018, 30, 8170–8178. [Google Scholar] [CrossRef]
- Mir, W.J.; Jagadeeswararao, M.; Das, S.; Nag, A. Colloidal Mn-Doped Cesium Lead Halide Perovskite Nanoplatelets. ACS Energy Lett. 2017, 2, 537–543. [Google Scholar] [CrossRef]
- Jellicoe, T.C.; Richter, J.M.; Glass, H.F.J.; Tabachnyk, M.; Brady, R.; Dutton, S.E.; Rao, A.; Friend, R.H.; Credgington, D.; Greenham, N.C.; et al. Synthesis and Optical Properties of Lead-Free Cesium Tin Halide Perovskite Nanocrystals. J. Am. Chem. Soc. 2016, 138, 2941–2944. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Deng, H.; Farooq, U.; Yang, X.; Khan, J.; Tang, J.; Song, H. High Quantum Yield Blue Emission from Lead-Free Inorganic Antimony Halide Perovskite Colloidal Quantum Dots. ACS Nano 2017, 11, 9294–9302. [Google Scholar] [CrossRef]
- Creutz, S.E.; Crites, E.N.; De Siena, M.C.; Gamelin, D.R. Colloidal Nanocrystals of Lead-Free Double-Perovskite (Elpasolite) Semiconductors: Synthesis and Anion Exchange To Access New Materials. Nano Lett. 2018, 18, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, R.; Gold-Parker, A.; Leijtens, T.; Conings, B.; Babayigit, A.; Boyen, H.-G.; Toney, M.F.; McGehee, M.D. Band Gap Tuning via Lattice Contraction and Octahedral Tilting in Perovskite Materials for Photovoltaics. J. Am. Chem. Soc. 2017, 139, 11117–11124. [Google Scholar] [CrossRef] [PubMed]
- Byranvand, M.M.; Otero-Martínez, C.; Ye, J.; Zuo, W.; Manna, L.; Saliba, M.; Hoye, R.L.Z.; Polavarapu, L. Recent Progress in Mixed A-Site Cation Halide Perovskite Thin-Films and Nanocrystals for Solar Cells and Light-Emitting Diodes. Adv. Opt. Mater. 2022, 10, 2200423. [Google Scholar] [CrossRef]
- Hazarika, A.; Zhao, Q.; Gaulding, E.A.; Christians, J.A.; Dou, B.; Marshall, A.R.; Moot, T.; Berry, J.J.; Johnson, J.C.; Luther, J.M. Perovskite Quantum Dot Photovoltaic Materials beyond the Reach of Thin Films: Full-Range Tuning of A-Site Cation Composition. ACS Nano 2018, 12, 10327–10337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yates, J.T., Jr. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Yang, M.-Z.; Chen, B.-X.; Wang, X.-D.; Chen, H.-Y.; Kuang, D.-B.; Su, C.-Y. A CsPbBr3 Perovskite Quantum Dot/Graphene Oxide Composite for Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 5660–5663. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Verhaeghe, D.; Weng, B.; Ghosh, B.; Zhang, H.; Hofkens, J.; Steele, J.A.; Roeffaers, M.B.J. Metal Halide Perovskite Based Heterojunction Photocatalysts. Angew. Chem. Int. Ed. 2022, 61, e202203261. [Google Scholar] [CrossRef]
- Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A.A. Heterojunction Photocatalysts. Adv. Mater. 2017, 29, 1601694. [Google Scholar] [CrossRef]
- Bard, A.J. Photoelectrochemistry and heterogeneous photo-catalysis at semiconductors. J. Photochem. 1979, 10, 59–75. [Google Scholar] [CrossRef]
- Schünemann, S.; van Gastel, M.; Tüysüz, H. A CsPbBr3/TiO2 Composite for Visible-Light-Driven Photocatalytic Benzyl Alcohol Oxidation. ChemSusChem 2018, 11, 2057–2061. [Google Scholar] [CrossRef]
- Xu, F.; Meng, K.; Cheng, B.; Wang, S.; Xu, J.; Yu, J. Unique S-scheme heterojunctions in self-assembled TiO2/CsPbBr3 hybrids for CO2 photoreduction. Nat. Commun. 2020, 11, 4613. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lin, Y.; Sun, Y.; Beard, M.C.; Yan, Y. Lead-Halide Perovskites for Photocatalytic α-Alkylation of Aldehydes. J. Am. Chem. Soc. 2019, 141, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lu, H.; Zhu, X.; Lin, Y.; Beard, M.C.; Yan, Y.; Chen, X. Ultrafast Reaction Mechanisms in Perovskite Based Photocatalytic C–C Coupling. ACS Energy Lett. 2020, 5, 566–571. [Google Scholar] [CrossRef]
- Rosa-Pardo, I.; Casadevall, C.; Schmidt, L.; Claros, M.; Galian, R.E.; Lloret-Fillol, J.; Pérez-Prieto, J. The synergy between the CsPbBr3 nanoparticle surface and the organic ligand becomes manifest in a demanding carbon–carbon coupling reaction. Chem. Commun. 2020, 56, 5026–5029. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhu, H.; Hills-Kimball, K.; Cai, T.; Shi, W.; Wei, Z.; Yang, H.; Candler, Y.; Wang, P.; He, J.; et al. Stereoselective C–C Oxidative Coupling Reactions Photocatalyzed by Zwitterionic Ligand Capped CsPbBr3 Perovskite Quantum Dots. Angew. Chem. Int. Ed. 2020, 59, 22563–22569. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Maneiro, N.; Sun, K.; López-Fernández, I.; Gómez-Graña, S.; Müller-Buschbaum, P.; Polavarapu, L. Ligand Chemistry of Inorganic Lead Halide Perovskite Nanocrystals. ACS Energy Lett. 2023, 8, 1152–1191. [Google Scholar] [CrossRef]
- Mir, W.J.; Alamoudi, A.; Yin, J.; Yorov, K.E.; Maity, P.; Naphade, R.; Shao, B.; Wang, J.; Lintangpradipto, M.N.; Nematulloev, S.; et al. Lecithin Capping Ligands Enable Ultrastable Perovskite-Phase CsPbI3 Quantum Dots for Rec. 2020 Bright-Red Light-Emitting Diodes. J. Am. Chem. Soc. 2022, 144, 13302–13310. [Google Scholar] [CrossRef]
- Manna, A.; Dinda, T.K.; Ghosh, S.; Mal, P. CsPbBr3 in the Activation of the C–Br Bond of CBrX3 (X = Cl, Br) under Sunlight. Chem. Mater. 2023, 35, 628–637. [Google Scholar] [CrossRef]
- Shi, T.; Sun, K.; Chen, X.-L.; Zhang, Z.-X.; Huang, X.-Q.; Peng, Y.-Y.; Qu, L.-B.; Yu, B. Recyclable Perovskite as Heterogeneous Photocatalyst for Aminomethylation of Imidazo-Fused Heterocycles. Adv. Synth. Catal. 2020, 362, 2143–2149. [Google Scholar] [CrossRef]
- Rohani, S.; Ziarati, A.; Ziarani, G.M.; Badiei, A.; Burgi, T. Engineering of highly active Au/Pd supported on hydrogenated urchin-like yolk@shell TiO2 for visible light photocatalytic Suzuki coupling. Catal. Sci. Technol. 2019, 9, 3820–3827. [Google Scholar] [CrossRef]
- Jiao, Z.; Zhai, Z.; Guo, X.; Guo, X.-Y. Visible-Light-Driven Photocatalytic Suzuki–Miyaura Coupling Reaction on Mott–Schottky-type Pd/SiC Catalyst. J. Phys. Chem. C 2015, 119, 3238–3243. [Google Scholar] [CrossRef]
- Raza, F.; Yim, D.; Park, J.H.; Kim, H.-I.; Jeon, S.-J.; Kim, J.-H. Structuring Pd Nanoparticles on 2H-WS2 Nanosheets Induces Excellent Photocatalytic Activity for Cross-Coupling Reactions under Visible Light. J. Am. Chem. Soc. 2017, 139, 14767–14774. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Liu, Y.; Wang, J.; Tan, X.; Sun, H.; Liu, S.; Wang, S. Temperature dependent photocatalysis of g-C3N4, TiO2 and ZnO: Differences in photoactive mechanism. J. Colloid Interface Sci. 2018, 532, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, P.; Zhang, D.; Qiao, W.; Wang, D.; Yang, X.; Wen, X.; Rummeli, M.H.; Niemantsverdriet, H.; Lewis, J.P.; et al. Rationally Designed Metal Cocatalyst for Selective Photosynthesis of Bibenzyls via Dehalogenative C–C Homocoupling. ACS Catal. 2021, 11, 4338–4348. [Google Scholar] [CrossRef]
- Wang, C.; Weng, B.; Keshavarz, M.; Yang, M.-Q.; Huang, H.; Ding, Y.; Lai, F.; Aslam, I.; Jin, H.; Romolini, G.; et al. Photothermal Suzuki Coupling Over a Metal Halide Perovskite/Pd Nanocube Composite Catalyst. ACS Appl. Mater. Interfaces 2022, 14, 17185–17194. [Google Scholar] [CrossRef]
- Li, X.-H.; Baar, M.; Blechert, S.; Antonietti, M. Facilitating room-temperature Suzuki coupling reaction with light: Mott-Schottky photocatalyst for C-C-coupling. Sci. Rep. 2013, 3, 1743. [Google Scholar] [CrossRef]
- Hong, Z.; Chong, W.K.; Ng, A.Y.R.; Li, M.; Ganguly, R.; Sum, T.C.; Soo, H.S. Hydrophobic Metal Halide Perovskites for Visible-Light Photoredox C–C Bond Cleavage and Dehydrogenation Catalysis. Angew. Chem. Int. Ed. 2019, 58, 3456–3460. [Google Scholar] [CrossRef]
- Zou, S.; Liu, Y.; Li, J.; Liu, C.; Feng, R.; Jiang, F.; Li, Y.; Song, J.; Zeng, H.; Hong, M.; et al. Stabilizing Cesium Lead Halide Perovskite Lattice through Mn(II) Substitution for Air-Stable Light-Emitting Diodes. J. Am. Chem. Soc. 2017, 139, 11443–11450. [Google Scholar] [CrossRef]
- Mu, Y.-F.; Zhang, W.; Guo, X.-X.; Dong, G.-X.; Zhang, M.; Lu, T.-B. Water-Tolerant Lead Halide Perovskite Nanocrystals as Efficient Photocatalysts for Visible-Light-Driven CO2 Reduction in Pure Water. ChemSusChem 2019, 12, 4769–4774. [Google Scholar] [CrossRef]
- Tang, C.; Chen, C.; Xu, W.; Xu, L. Design of doped cesium lead halide perovskite as a photo-catalytic CO2 reduction catalyst. J. Mater. Chem. A 2019, 7, 6911–6919. [Google Scholar] [CrossRef]
- Shyamal, S.; Dutta, S.K.; Pradhan, N. Doping Iron in CsPbBr3 Perovskite Nanocrystals for Efficient and Product Selective CO2 Reduction. J. Phys. Chem. Lett. 2019, 10, 7965–7969. [Google Scholar] [CrossRef]
- Martin, J.S.; Zeng, X.; Chen, X.; Miller, C.; Han, C.; Lin, Y.; Yamamoto, N.; Wang, X.; Yazdi, S.; Yan, Y.; et al. A Nanocrystal Catalyst Incorporating a Surface Bound Transition Metal to Induce Photocatalytic Sequential Electron Transfer Events. J. Am. Chem. Soc. 2021, 143, 11361–11369. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Deng, X.; Dodekatos, G.; Tüysüz, H. Photocatalytic Polymerization of 3,4-Ethylenedioxythiophene over Cesium Lead Iodide Perovskite Quantum Dots. J. Am. Chem. Soc. 2017, 139, 12267–12273. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shu, Q.; Du, Q.; Dai, Y.; Zhao, S.; Zhang, J.; Li, L.; Chen, K. Surface Modification for Improving the Photocatalytic Polymerization of 3,4-Ethylenedioxythiophene over Inorganic Lead Halide Perovskite Quantum Dots. ACS Appl. Mater. Interfaces 2020, 12, 451–460. [Google Scholar] [CrossRef]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The Role of Thiols and Disulfides on Protein Stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.L.; Powis, G.; Hornsveld, M.; Dansen, T.B.; Maulucci, G.; Bačić, G.; Bridal, L.; Schmidt, H.H.; Tavitian, B.; Viel, T.; et al. Clinically Evaluated Cancer Drugs Inhibiting Redox Signaling. Antioxidants Redox Signal. 2017, 26, 262–273. [Google Scholar] [CrossRef]
- Berkmen, M. Production of disulfide-bonded proteins in Escherichia coli. Protein Expr. Purif. 2012, 82, 240–251. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Abbruzzese, J.; Dragovich, T.; Kirkpatrick, L.; Guillen, J.M.; Baker, A.F.; Pestano, L.A.; Green, S.; Von Hoff, D.D. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother. Pharmacol. 2011, 67, 503–509. [Google Scholar] [CrossRef]
- Huang, H.; Yuan, H.; Janssen, K.P.F.; Solís-Fernández, G.; Wang, Y.; Tan, C.Y.X.; Jonckheere, D.; Debroye, E.; Long, J.; Hendrix, J.; et al. Efficient and Selective Photocatalytic Oxidation of Benzylic Alcohols with Hybrid Organic–Inorganic Perovskite Materials. ACS Energy Lett. 2018, 3, 755–759. [Google Scholar] [CrossRef]
- McClelland, K.P.; Weiss, E.A. Selective Photocatalytic Oxidation of Benzyl Alcohol to Benzaldehyde or C–C Coupled Products by Visible-Light-Absorbing Quantum Dots. ACS Appl. Energy Mater. 2019, 2, 92–96. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, T.; Han, F.; Yang, Q.; Li, B. Synthesis of Ni modified Au@CdS core–shell nanostructures for enhancing photocatalytic coproduction of hydrogen and benzaldehyde under visible light. J. Colloid Interface Sci. 2022, 606, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Kang, M.J.; Park, M.; Kim, K.-j.; Lee, H.; Kim, H.S. Selective photocatalytic conversion of benzyl alcohol to benzaldehyde or deoxybenzoin over ion-exchanged CdS. Appl. Catal. B Environ. 2022, 304, 120967. [Google Scholar] [CrossRef]
- Ramesh, A.; Da, C.T.; Manigandan, R.; Bhargav, P.B.; Nguyen-Le, M.-T. Selectivity oxidation of benzyl alcohol using mesoporous g-C3N4 catalysts prepared by hard template method. Colloid Interface Sci. Commun. 2022, 48, 100608. [Google Scholar] [CrossRef]
- Huang, H.; Yuan, H.; Zhao, J.; Solís-Fernández, G.; Zhou, C.; Seo, J.W.; Hendrix, J.; Debroye, E.; Steele, J.A.; Hofkens, J.; et al. C(sp3)–H Bond Activation by Perovskite Solar Photocatalyst Cell. ACS Energy Lett. 2019, 4, 203–208. [Google Scholar] [CrossRef]
- Dai, Y.; Tüysüz, H. Lead-Free Cs3Bi2Br9 Perovskite as Photocatalyst for Ring-Opening Reactions of Epoxides. ChemSusChem 2019, 12, 2587–2592. [Google Scholar] [CrossRef]
- Wong, Y.-C.; De Andrew Ng, J.; Tan, Z.-K. Perovskite-Initiated Photopolymerization for Singly Dispersed Luminescent Nanocomposites. Adv. Mater. 2018, 30, 1800774. [Google Scholar] [CrossRef]
- Long, G.; Sabatini, R.; Saidaminov, M.I.; Lakhwani, G.; Rasmita, A.; Liu, X.; Sargent, E.H.; Gao, W. Chiral-perovskite optoelectronics. Nat. Rev. Mater. 2020, 5, 423–439. [Google Scholar] [CrossRef]
- Mishra, K.; Guyon, D.; San Martin, J.; Yan, Y. Chiral Perovskite Nanocrystals for Asymmetric Reactions: A Highly Enantioselective Strategy for Photocatalytic Synthesis of N–C Axially Chiral Heterocycles. J. Am. Chem. Soc. 2023, 145, 17242–17252. [Google Scholar] [CrossRef]
- Ghosh, I.; Shaikh, R.S.; König, B. Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 8544–8549. [Google Scholar] [CrossRef]
- Ravetz, B.D.; Pun, A.B.; Churchill, E.M.; Congreve, D.N.; Rovis, T.; Campos, L.M. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 2019, 565, 343–346. [Google Scholar] [CrossRef]
- Schulze, T.F.; Schmidt, T.W. Photochemical upconversion: Present status and prospects for its application to solar energy conversion. Energy Environ. Sci. 2015, 8, 103–125. [Google Scholar] [CrossRef]
- Schmidt, T.W.; Castellano, F.N. Photochemical Upconversion: The Primacy of Kinetics. J. Phys. Chem. Lett. 2014, 5, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Meier, F.; Zhao, D.; Abe, Y.; Gao, Y.; Chen, B.; Salim, T.; Chia, E.E.M.; Qiao, X.; Deibel, C.; et al. Efficient Room-Temperature Phosphorescence from Organic–Inorganic Hybrid Perovskites by Molecular Engineering. Adv. Mater. 2018, 30, 1707621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, J.; Wu, S.; Wang, Z.; Wu, W.; Ma, J.; Guo, S.; Huang, L. Intramolecular RET Enhanced Visible Light-Absorbing Bodipy Organic Triplet Photosensitizers and Application in Photooxidation and Triplet–Triplet Annihilation Upconversion. J. Am. Chem. Soc. 2013, 135, 10566–10578. [Google Scholar] [CrossRef] [PubMed]
- Moor, K.; Kim, J.-H.; Snow, S.; Kim, J.-H. [C70] Fullerene-sensitized triplet–triplet annihilation upconversion. Chem. Commun. 2013, 49, 10829–10831. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guo, H.; Wu, W.; Ji, S.; Zhao, J. Organic Triplet Sensitizer Library Derived from a Single Chromophore (BODIPY) with Long-Lived Triplet Excited State for Triplet–Triplet Annihilation Based Upconversion. J. Org. Chem. 2011, 76, 7056–7064. [Google Scholar] [CrossRef] [PubMed]
- Yanai, N.; Kimizuka, N. New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption. Acc. Chem. Res. 2017, 50, 2487–2495. [Google Scholar] [CrossRef] [PubMed]
- Mongin, C.; Garakyaraghi, S.; Razgoniaeva, N.; Zamkov, M.; Castellano, F.N. Direct observation of triplet energy transfer from semiconductor nanocrystals. Science 2016, 351, 369–372. [Google Scholar] [CrossRef]
- Wu, M.; Congreve, D.N.; Wilson, M.W.B.; Jean, J.; Geva, N.; Welborn, M.; Van Voorhis, T.; Bulović, V.; Bawendi, M.G.; Baldo, M.A. Solid-state infrared-to-visible upconversion sensitized by colloidal nanocrystals. Nat. Photonics 2016, 10, 31–34. [Google Scholar] [CrossRef]
- Mase, K.; Okumura, K.; Yanai, N.; Kimizuka, N. Triplet sensitization by perovskite nanocrystals for photon upconversion. Chem. Commun. 2017, 53, 8261–8264. [Google Scholar] [CrossRef]
- Han, Y.; Luo, X.; Lai, R.; Li, Y.; Liang, G.; Wu, K. Visible-Light-Driven Sensitization of Naphthalene Triplets Using Quantum-Confined CsPbBr3 Nanocrystals. J. Phys. Chem. Lett. 2019, 10, 1457–1463. [Google Scholar] [CrossRef]
- DuBose, J.T.; Kamat, P.V. Energy Versus Electron Transfer: Managing Excited-State Interactions in Perovskite Nanocrystal–Molecular Hybrids. Chem. Rev. 2022, 122, 12475–12494. [Google Scholar] [CrossRef]
- Luo, X.; Lai, R.; Li, Y.; Han, Y.; Liang, G.; Liu, X.; Ding, T.; Wang, J.; Wu, K. Triplet Energy Transfer from CsPbBr3 Nanocrystals Enabled by Quantum Confinement. J. Am. Chem. Soc. 2019, 141, 4186–4190. [Google Scholar] [CrossRef]
- Liu, M.; Xia, P.; Zhao, G.; Nie, C.; Gao, K.; He, S.; Wang, L.; Wu, K. Energy-Transfer Photocatalysis Using Lead Halide Perovskite Nanocrystals: Sensitizing Molecular Isomerization and Cycloaddition. Angew. Chem. Int. Ed. 2022, 61, e202208241. [Google Scholar] [CrossRef]
- Lin, Y.; Avvacumova, M.; Zhao, R.; Chen, X.; Beard, M.C.; Yan, Y. Triplet Energy Transfer from Lead Halide Perovskite for Highly Selective Photocatalytic 2 + 2 Cycloaddition. ACS Appl. Mater. Interfaces 2022, 14, 25357–25365. [Google Scholar] [CrossRef] [PubMed]
- Coperet, C. C–H Bond Activation and Organometallic Intermediates on Isolated Metal Centers on Oxide Surfaces. Chem. Rev. 2010, 110, 656–680. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Chen, J.-S.; Wang, X.; Sun, J.; Antonietti, M. Metal-Free Activation of Dioxygen by Graphene/g-C3N4 Nanocomposites: Functional Dyads for Selective Oxidation of Saturated Hydrocarbons. J. Am. Chem. Soc. 2011, 133, 8074–8077. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, L.; Tiruvalam, R.; Rahim, M.H.A.; bin Saiman, M.I.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; et al. Solvent-Free Oxidation of Primary Carbon-Hydrogen Bonds in Toluene Using Au-Pd Alloy Nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Partenheimer, W. Methodology and scope of metal/bromide autoxidation of hydrocarbons. Catal. Today 1995, 23, 69–158. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.; Gao, F.; Luo, D. g-C3N4-Stabilised Organic–Inorganic Halide Perovskites for Efficient Photocatalytic Selective Oxidation of Benzyl Alcohol. Catalysts 2021, 11, 505. [Google Scholar] [CrossRef]
- Zhao, Y.; Dai, Y.; Wang, Q.; Dong, Y.; Song, T.; Mudryi, A.; Chen, Q.; Li, Y. Anions-Exchange-Induced Efficient Carrier Transport at CsPbBrxCl3-x/TiO2 Interface for Photocatalytic Activation of C(sp3)–H bond in Toluene Oxidation. ChemCatChem 2021, 13, 2592–2598. [Google Scholar] [CrossRef]
- Zhu, E.; Zhao, Y.; Dai, Y.; Wang, Q.; Dong, Y.; Chen, Q.; Li, Y. Heterojunction-Type Photocatalytic System Based on Inorganic Halide Perovskite CsPbBr3 Chin. J. Chem. 2020, 38, 1718–1722. [Google Scholar] [CrossRef]
- Chen, T.; Li, M.; Shen, L.; Roeffaers, M.B.J.; Weng, B.; Zhu, H.; Chen, Z.; Yu, D.; Pan, X.; Yang, M.-Q.; et al. Photocatalytic Anaerobic Oxidation of Aromatic Alcohols Coupled with H2 Production Over CsPbBr3/GO-Pt Catalysts. Front. Chem. 2022, 10, 833784. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, H.; Weng, B.; Verhaeghe, D.; Keshavarz, M.; Jin, H.; Liu, B.; Xie, H.; Ding, Y.; Gao, Y.; et al. Planar heterojunction boosts solar-driven photocatalytic performance and stability of halide perovskite solar photocatalyst cell. Appl. Catal. B Environ. 2022, 301, 120760. [Google Scholar] [CrossRef]
- Yi, J.; Ke, S.; Lu, S.; Weng, B.; Shen, L.; Yang, X.; Xue, H.; Yang, M.-Q.; Qian, Q. High-efficiency visible-light-driven oxidation of primary C–H bonds in toluene over a CsPbBr3 perovskite supported by hierarchical TiO2 nanoflakes. Nanoscale 2023, 15, 14584–14594. [Google Scholar] [CrossRef]
- Clennan, E.L.; Pace, A. Advances in singlet oxygen chemistry. Tetrahedron 2005, 61, 6665–6691. [Google Scholar] [CrossRef]
- Alberti, M.N.; Orfanopoulos, M. Unraveling the Mechanism of the Singlet Oxygen Ene Reaction: Recent Computational and Experimental Approaches. Chem.–Eur. J. 2010, 16, 9414–9421. [Google Scholar] [CrossRef]
- Ogilby, P.R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. [Google Scholar] [CrossRef]
- Camussi, I.; Mannucci, B.; Speltini, A.; Profumo, A.; Milanese, C.; Malavasi, L.; Quadrelli, P. g-C3N4—Singlet Oxygen Made Easy for Organic Synthesis: Scope and Limitations. ACS Sustain. Chem. Eng. 2019, 7, 8176–8182. [Google Scholar] [CrossRef]
- Corti, M.; Chiara, R.; Romani, L.; Mannucci, B.; Malavasi, L.; Quadrelli, P. g-C3N4/metal halide perovskite composites as photocatalysts for singlet oxygen generation processes for the preparation of various oxidized synthons. Catal. Sci. Technol. 2021, 11, 2292–2298. [Google Scholar] [CrossRef]
- Dai, Y.; Tüysüz, H. Rapid Acidic Media Growth of Cs3Bi2Br9 Halide Perovskite Platelets for Photocatalytic Toluene Oxidation. Sol. RRL 2021, 5, 2100265. [Google Scholar] [CrossRef]
- Wang, C.; Weng, B.; Liao, Y.; Liu, B.; Keshavarz, M.; Ding, Y.; Huang, H.; Verhaeghe, D.; Steele, J.A.; Feng, W.; et al. Simultaneous photocatalytic H2 generation and organic synthesis over crystalline–amorphous Pd nanocube decorated Cs3Bi2Br9. Chem. Commun. 2022, 58, 10691–10694. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ding, Y.; Liu, B.; Weng, B.; Hofkens, J.; Roeffaers, M.B.J. Crystal structure engineering of metal halide perovskites for photocatalytic organic synthesis. Chem. Commun. 2023, 59, 3122–3125. [Google Scholar] [CrossRef]
- Sun, Q.; Ye, W.; Wei, J.; Li, L.; Wang, J.; He, J.-H.; Lu, J.-M. Lead-free perovskite Cs3Bi2Br9 heterojunctions for highly efficient and selective photocatalysis under mild conditions. J. Alloys Compd. 2022, 893, 162326. [Google Scholar] [CrossRef]
- Eklund, P.; Beckers, M.; Jansson, U.; Högberg, H.; Hultman, L. The Mn+1AXn phases: Materials science and thin-film processing. Thin Solid Films 2010, 518, 1851–1878. [Google Scholar] [CrossRef]
- Naguib, M.; Mashtalir, O.; Carle, J.; Presser, V.; Lu, J.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Transition Metal Carbides. ACS Nano 2012, 6, 1322–1331. [Google Scholar] [CrossRef]
- Pandey, P.; Sengupta, A.; Parmar, S.; Bansode, U.; Gosavi, S.; Swarnkar, A.; Muduli, S.; Mohite, A.D.; Ogale, S. CsPbBr3–Ti3C2Tx MXene QD/QD Heterojunction: Photoluminescence Quenching, Charge Transfer, and Cd Ion Sensing Application. ACS Appl. Nano Mater. 2020, 3, 3305–3314. [Google Scholar] [CrossRef]
- Li, Q.; Song, T.; Zhang, Y.; Wang, Q.; Yang, Y. Boosting Photocatalytic Activity and Stability of Lead-Free Cs3Bi2Br9 Perovskite Nanocrystals via In Situ Growth on Monolayer 2D Ti3C2Tx MXene for C–H Bond Oxidation. ACS Appl. Mater. Interfaces 2021, 13, 27323–27333. [Google Scholar] [CrossRef]
- Wen, J.; Xie, J.; Chen, X.; Li, X. A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 2017, 391, 72–123. [Google Scholar] [CrossRef]
- Xu, J.; Brenner, T.J.K.; Chabanne, L.; Neher, D.; Antonietti, M.; Shalom, M. Liquid-Based Growth of Polymeric Carbon Nitride Layers and Their Use in a Mesostructured Polymer Solar Cell with Voc Exceeding 1 V. J. Am. Chem. Soc. 2014, 136, 13486–13489. [Google Scholar] [CrossRef]
- Xu, J.; Wang, G.; Fan, J.; Liu, B.; Cao, S.; Yu, J. g-C3N4 modified TiO2 nanosheets with enhanced photoelectric conversion efficiency in dye-sensitized solar cells. J. Power Sources 2015, 274, 77–84. [Google Scholar] [CrossRef]
- Shanker, G.S.; Markad, G.B.; Jagadeeswararao, M.; Bansode, U.; Nag, A. Colloidal Nanocomposite of TiN and N-Doped Few-Layer Graphene for Plasmonics and Electrocatalysis. ACS Energy Lett. 2017, 2, 2251–2256. [Google Scholar] [CrossRef]
- Bai, Z.-J.; Mao, Y.; Wang, B.-H.; Chen, L.; Tian, S.; Hu, B.; Li, Y.-J.; Au, C.-T.; Yin, S.-F. Tuning photocatalytic performance of Cs3Bi2Br9 perovskite by g-C3N4 for C(sp3)—H bond activation. Nano Res. 2023, 16, 6104–6112. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Z.; Huang, H.; Liu, Y.; Zou, J.; Shen, S.; Yan, J.; Zhang, J.; Zhuang, Z.; Luo, Z.; et al. Synergistic surface activation during photocatalysis on perovskite derivative sites in heterojunction. Appl. Catal. B Environ. 2023, 323, 122146. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Zhou, Z.-C.; Dong, Y.-J.; Zhang, L.; Chen, H.-Y.; Kuang, D.-B. Constructing a Cs3Sb2Br9/g-C3N4 Hybrid for Photocatalytic Aromatic C(sp3)-H Bond Activation. Sol. RRL 2021, 5, 2100559. [Google Scholar] [CrossRef]
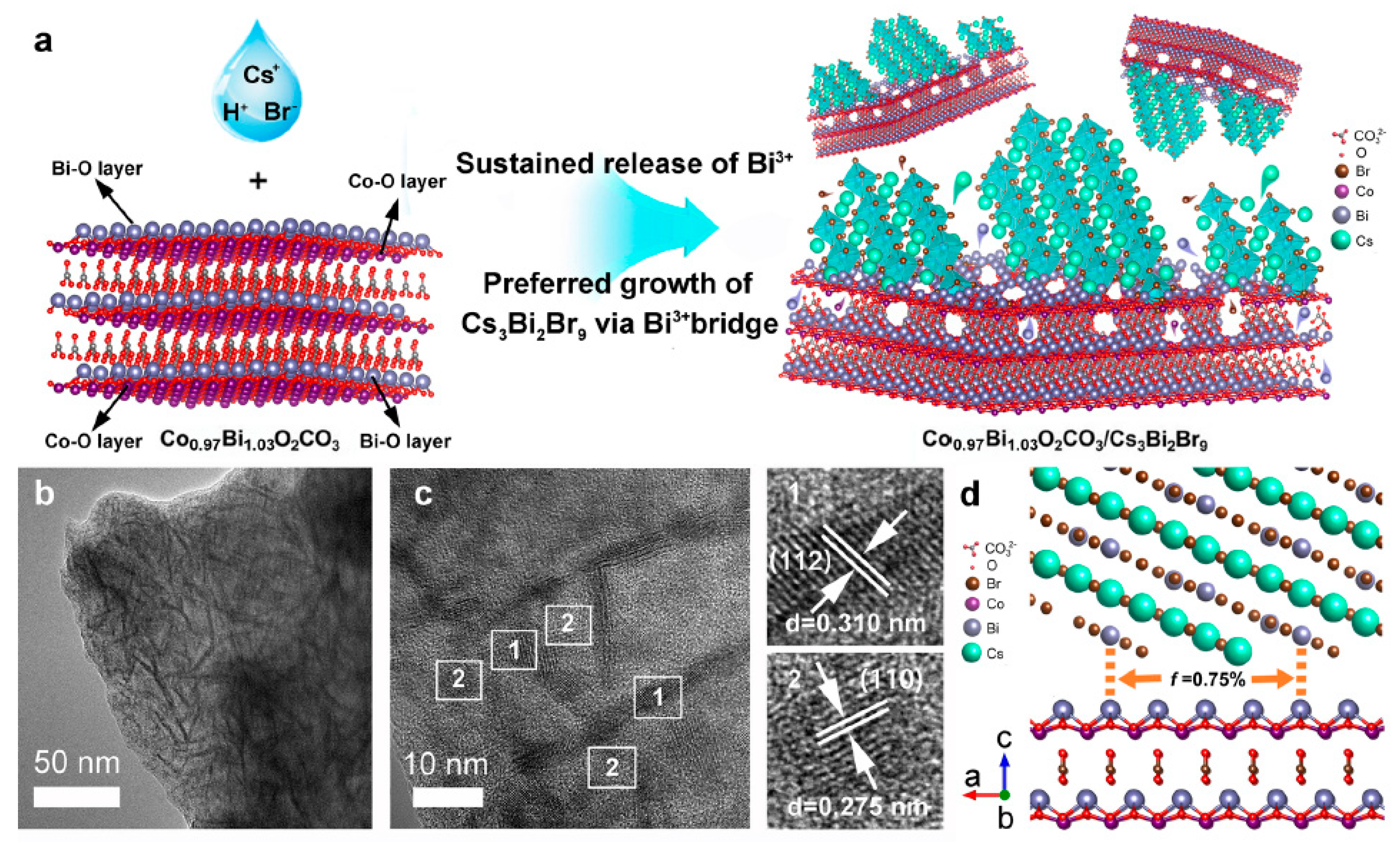
- Teng, Y.; Chen, J.-H.; Huang, Y.-H.; Zhou, Z.-C.; Wang, X.-D.; Kuang, D.-B.; Chen, H.-Y. Atom-triggered epitaxial growth of Bi-based perovskite heterojunctions for promoting interfacial charge transfer. Appl. Catal. B Environ. 2023, 335, 122889. [Google Scholar] [CrossRef]
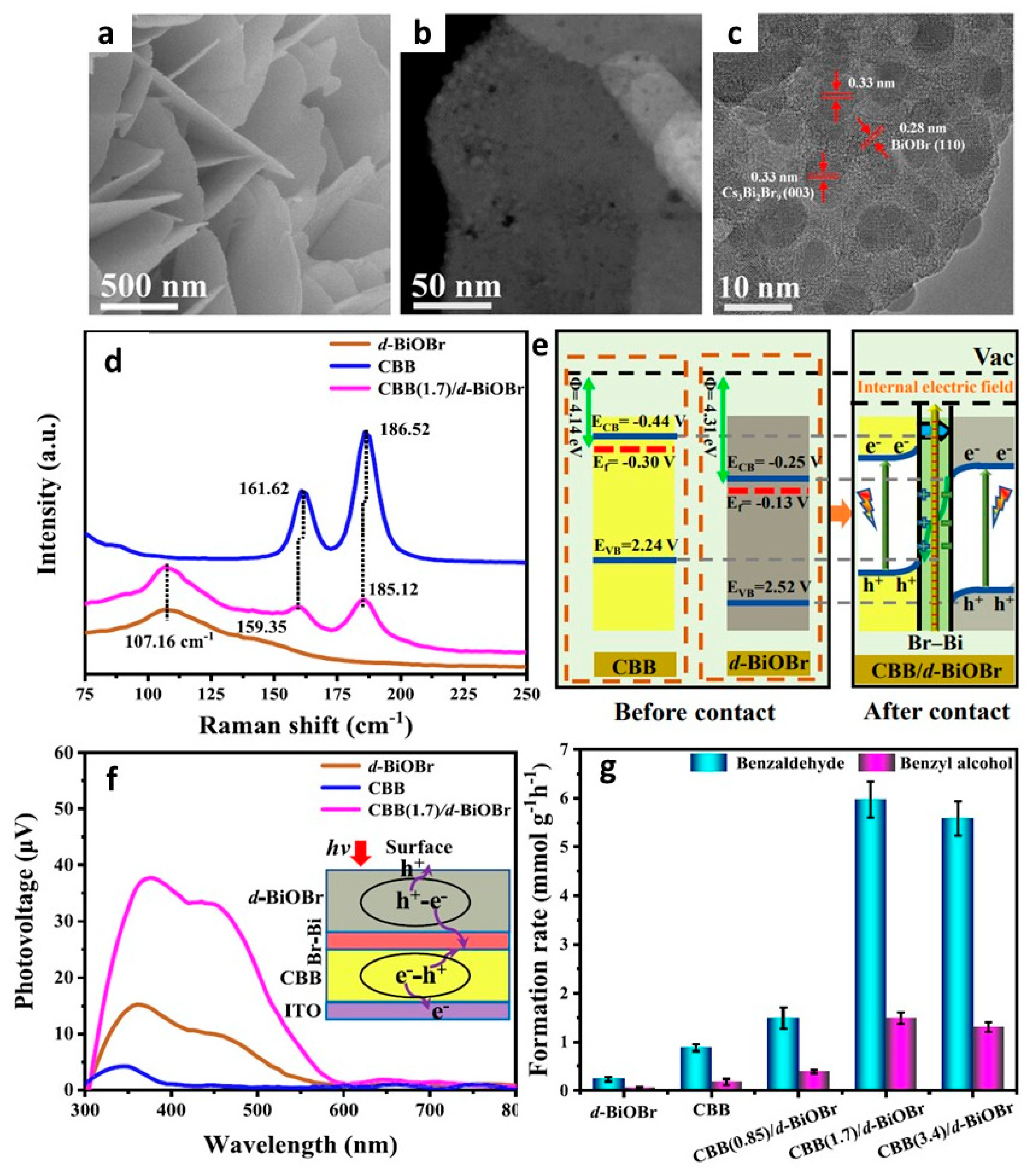
- Bai, Z.-J.; Tian, S.; Zeng, T.-Q.; Chen, L.; Wang, B.-H.; Hu, B.; Wang, X.; Zhou, W.; Pan, J.-B.; Shen, S.; et al. Cs3Bi2Br9 Nanodots Stabilized on Defective BiOBr Nanosheets by Interfacial Chemical Bonding: Modulated Charge Transfer for Photocatalytic C(sp3)–H Bond Activation. ACS Catal. 2022, 12, 15157–15167. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, J.; Du, Y.; Zhou, C.; Zhang, M.; Wang, Z.; Weng, Y.; Long, J.; Hofkens, J.; Steele, J.A.; et al. Direct Z-Scheme Heterojunction of Semicoherent FAPbBr3/Bi2WO6 Interface for Photoredox Reaction with Large Driving Force. ACS Nano 2020, 14, 16689–16697. [Google Scholar] [CrossRef]
- Wang, W.; Huang, H.; Ke, X.; Liu, X.; Yang, S.; Wang, K.; Huang, L.; Tu, C.; Zheng, Z.; Luo, D.; et al. Morphology- and size-dependent FAPbBr3/WO3 Z-scheme photocatalysts for the controllable photo-oxidation of benzyl alcohol. Mater. Des. 2022, 215, 110502. [Google Scholar] [CrossRef]
- Mu, Y.-F.; Zhang, C.; Zhang, M.-R.; Zhang, W.; Zhang, M.; Lu, T.-B. Direct Z-Scheme Heterojunction of Ligand-Free FAPbBr3/α-Fe2O3 for Boosting Photocatalysis of CO2 Reduction Coupled with Water Oxidation. ACS Appl. Mater. Interfaces 2021, 13, 22314–22322. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, H.-Y.; Li, J.-Y.; Liao, J.-F.; Zhang, H.-H.; Wang, X.-D.; Kuang, D.-B. Z-Scheme 2D/2D Heterojunction of CsPbBr3/Bi2WO6 for Improved Photocatalytic CO2 Reduction. Adv. Funct. Mater. 2020, 30, 2004293. [Google Scholar] [CrossRef]
- Bai, Z.-J.; Tan, X.-P.; Chen, L.; Hu, B.; Tan, Y.-X.; Mao, Y.; Shen, S.; Guo, J.-K.; Au, C.-T.; Liang, Z.-W.; et al. Efficient photocatalytic toluene selective oxidation over Cs3Bi1.8Sb0.2Br9 Nanosheets: Enhanced charge carriers generation and C–H bond dissociation. Chem. Eng. Sci. 2022, 247, 116983. [Google Scholar] [CrossRef]
- Goyal, A.; McKechnie, S.; Pashov, D.; Tumas, W.; van Schilfgaarde, M.; Stevanović, V. Origin of Pronounced Nonlinear Band Gap Behavior in Lead–Tin Hybrid Perovskite Alloys. Chem. Mater. 2018, 30, 3920–3928. [Google Scholar] [CrossRef]
- Shi, M.; Zhou, H.; Tian, W.; Yang, B.; Yang, S.; Han, K.; Li, R.; Li, C. Lead-free B-site bimetallic perovskite photocatalyst for efficient benzylic C–H bond activation. Cell Rep. Phys. Sci. 2021, 2, 100656. [Google Scholar] [CrossRef]
- Cui, Z.; Wu, Y.; Zhang, S.; Fu, H.; Chen, G.; Lou, Z.; Liu, X.; Zhang, Q.; Wang, Z.; Zheng, Z.; et al. Insight into a strategy to improve charge carrier migration in lead-free bismuth-based halide perovskite for efficient selective oxidation of thioanisole under visible light. Chem. Eng. J. 2023, 451, 138927. [Google Scholar] [CrossRef]
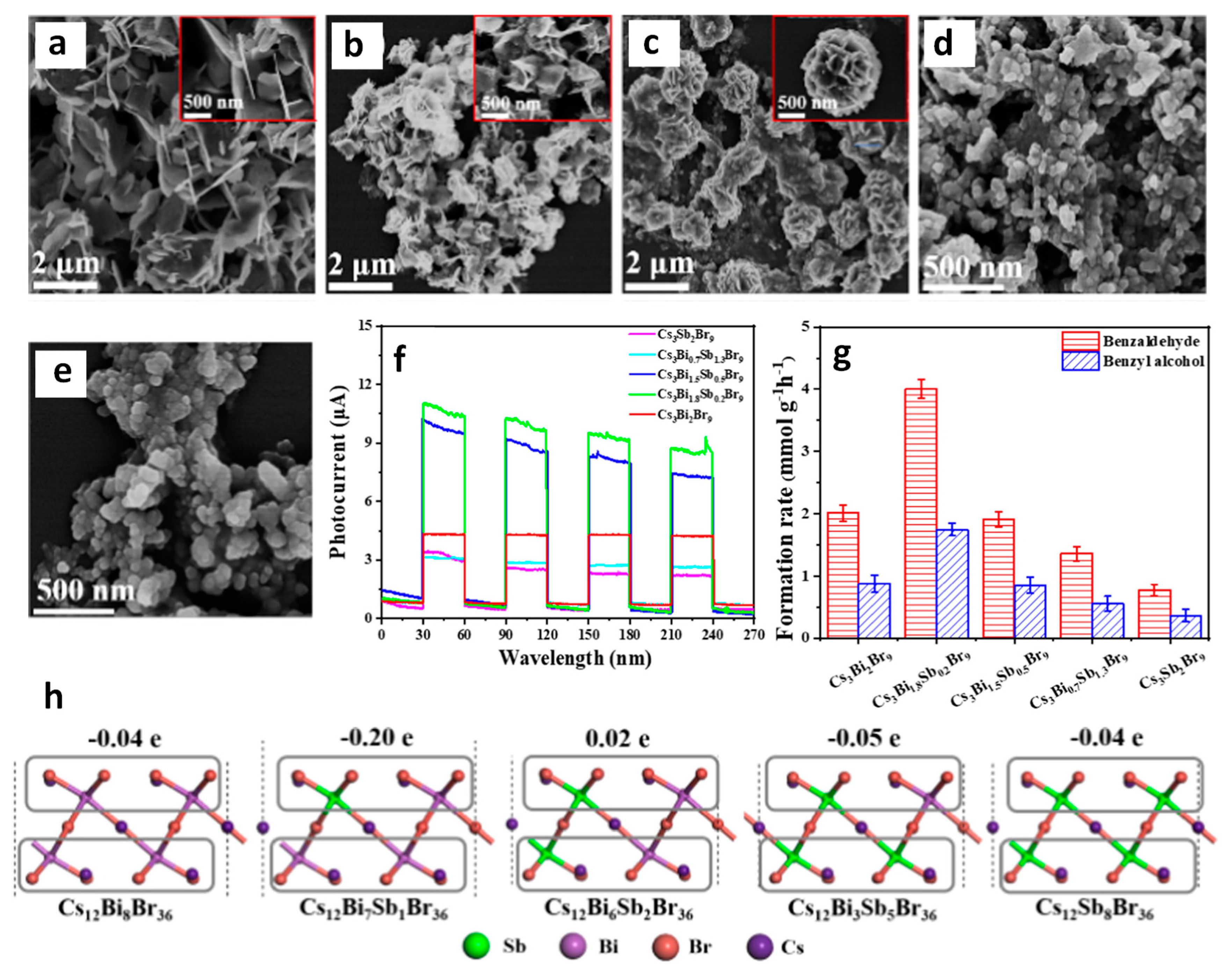
- Zhang, Z.; Yang, Y.; Wang, Y.; Yang, L.; Li, Q.; Chen, L.; Xu, D. Revealing the A-Site Effect of Lead-Free A3Sb2Br9 Perovskite in Photocatalytic C(sp3)–H Bond Activation. Angew. Chem. Int. Ed. 2020, 59, 18136–18139. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Poidevin, C.; Ochoa-Hernández, C.; Auer, A.A.; Tüysüz, H. A Supported Bismuth Halide Perovskite Photocatalyst for Selective Aliphatic and Aromatic C–H Bond Activation. Angew. Chem. Int. Ed. 2020, 59, 5788–5796. [Google Scholar] [CrossRef]
- Shi, M.; Li, G.; Tian, W.; Jin, S.; Tao, X.; Jiang, Y.; Pidko, E.A.; Li, R.; Li, C. Understanding the Effect of Crystalline Structural Transformation for Lead-Free Inorganic Halide Perovskites. Adv. Mater. 2020, 32, 2002137. [Google Scholar] [CrossRef]
- Slavney, A.H.; Leppert, L.; Bartesaghi, D.; Gold-Parker, A.; Toney, M.F.; Savenije, T.J.; Neaton, J.B.; Karunadasa, H.I. Defect-Induced Band-Edge Reconstruction of a Bismuth-Halide Double Perovskite for Visible-Light Absorption. J. Am. Chem. Soc. 2017, 139, 5015–5018. [Google Scholar] [CrossRef] [PubMed]
- Du, K.-z.; Meng, W.; Wang, X.; Yan, Y.; Mitzi, D.B. Bandgap Engineering of Lead-Free Double Perovskite Cs2AgBiBr6 through Trivalent Metal Alloying. Angew. Chem. Int. Ed. 2017, 56, 8158–8162. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, Z.; Wu, C.; Zhang, Y.; Li, X.; Yu, W.; Yao, G.; Liu, S.; Shi, J.-j.; Liu, K.; et al. Extending Absorption of Cs2AgBiBr6 to Near-Infrared Region (≈1350 nm) with Intermediate Band. Adv. Funct. Mater. 2022, 32, 2109891. [Google Scholar] [CrossRef]
- Li, X.; Mai, H.; Cox, N.; Lu, J.; Wen, X.; Chen, D.; Caruso, R.A. Sb-Substituted Cs2AgBiBr6/g-C3N4 Composite for Photocatalytic C(sp3)–H Bond Activation in Toluene. Chem. Mater. 2023, 35, 3105–3114. [Google Scholar] [CrossRef]
- Song, J.; Zhang, C.; Zhang, H.; Dai, D.; Zhang, Q.; Wang, Z.; Zheng, Z.; Liu, Y.; Cheng, H.; Dai, Y.; et al. In situ growth of lead-free perovskite Cs2AgBiBr6 on a flexible ultrathin carbon nitride sheet for highly efficient photocatalytic benzylic C(sp3)–H bond activation. Chem. Eng. J. 2023, 453, 139748. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, N.; Tang, Z.-R.; Xu, Y.-J. Transforming CdS into an efficient visible light photocatalyst for selective oxidation of saturated primary C–H bonds under ambient conditions. Chem. Sci. 2012, 3, 2812–2822. [Google Scholar] [CrossRef]
- Cao, F.-L.; Wang, J.-G.; Lv, F.-J.; Zhang, D.-Q.; Huo, Y.-N.; Li, G.-S.; Li, H.-X.; Zhu, J. Photocatalytic oxidation of toluene to benzaldehyde over anatase TiO2 hollow spheres with exposed {001} facets. Catal. Commun. 2011, 12, 946–950. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Yuan, Q.; He, J.; Au, C.-T.; Yin, S.-F. A green and efficient photocatalytic route for the highly-selective oxidation of saturated alpha-carbon C–H bonds in aromatic alkanes over flower-like Bi2WO6. Chem. Commun. 2016, 52, 1274–1277. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Z.; Xin, X.; Zhang, J.; Feng, Y.; Lv, H. Selective Valorization of 5-Hydroxymethylfurfural to 2,5-Diformylfuran Using Atmospheric O2 and MAPbBr3 Perovskite under Visible Light. ACS Catal. 2020, 10, 14793–14800. [Google Scholar] [CrossRef]
- Yin, L.; Wu, Y.; Bao, X.; Liu, X.; Dai, D.; Zhang, M.; Wang, Z.; Zheng, Z.; Liu, Y.; Cheng, H.; et al. Lead-Free Perovskite Cs2SnBr6/rGO Composite for Photocatalytic Selective Oxidation of 5-Hydroxymethylfurfural to 2,5-Diformylfuran. Chem.–Eur. J. 2023, 29, e202300999. [Google Scholar] [CrossRef]
- Rosa-Pardo, I.; Zhu, D.; Cortés-Villena, A.; Prato, M.; De Trizio, L.; Manna, L.; Galian, R.E.; Pérez-Prieto, J. The Dark Side of Lead-Free Metal Halide Nanocrystals: Substituent-Modulated Photocatalytic Activity in Benzyl Bromide Reduction. ACS Energy Lett. 2023, 8, 2789–2798. [Google Scholar] [CrossRef]
- Guo, Y.; Lou, Y.; Chen, J.; Zhao, Y. Lead-Free Cs2AgSbCl6 Double Perovskite Nanocrystals for Effective Visible-Light Photocatalytic C–C Coupling Reactions. ChemSusChem 2022, 15, e202102334. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswararao, M.; Sim, K.M.; Lee, S.; Kang, M.; An, S.; Nam, G.-H.; Sim, H.R.; Oleiki, E.; Lee, G.; Chung, D.S. Stoichiometric Engineering of Cs2AgBiBr6 for Photomultiplication-Type Photodetectors. Chem. Mater. 2023, 35, 3095–3104. [Google Scholar] [CrossRef]
- Jing, J.; Chen, Z.; Feng, C.; Sun, M.; Hou, J. Transforming g-C3N4 from amphoteric to n-type semiconductor: The important role of p/n type on photoelectrochemical cathodic protection. J. Alloys Compd. 2021, 851, 156820. [Google Scholar] [CrossRef]
- Luo, J.; Dong, G.; Zhu, Y.; Yang, Z.; Wang, C. Switching of semiconducting behavior from n-type to p-type induced high photocatalytic NO removal activity in g-C3N4. Appl. Catal. B Environ. 2017, 214, 46–56. [Google Scholar] [CrossRef]
- Bera, S.; Pradhan, N. Perovskite Nanocrystal Heterostructures: Synthesis, Optical Properties, and Applications. ACS Energy Lett. 2020, 5, 2858–2872. [Google Scholar] [CrossRef]
- Kipkorir, A.; DuBose, J.; Cho, J.; Kamat, P.V. CsPbBr3–CdS heterostructure: Stabilizing perovskite nanocrystals for photocatalysis. Chem. Sci. 2021, 12, 14815–14825. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Pardo, I.; Ciccone, A.; Arenal, R.; Galian, R.E.; Pérez-Prieto, J. One-Pot Synthesis of Stable CsPbBr3@CsPb2Br5 Core–Shell Heteronanocrystals with Controlled Permeability to Halide Ions. Chem. Mater. 2023, 35, 7011–7019. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, C.; Huang, S.; Li, Z.; Kong, L.; Jin, L.; Wang, J.; Wu, K.; Li, L. Postsynthesis Phase Transformation for CsPbBr3/Rb4PbBr6 Core/Shell Nanocrystals with Exceptional Photostability. ACS Appl. Mater. Interfaces 2018, 10, 23303–23310. [Google Scholar] [CrossRef]
- Ruan, L.J.; Tang, B.; Ma, Y. Improving the Stability of CsPbBr3 Nanocrystals in Ethanol by Capping with PbBr2-Adlayers. J. Phys. Chem. C 2019, 123, 11959–11967. [Google Scholar] [CrossRef]
- Cai, J.; Gu, K.; Zhu, Y.; Zhu, J.; Wang, Y.; Shen, J.; Trinchi, A.; Li, C.; Wei, G. Highly stable CsPbBr3@SiO2 nanocomposites prepared via confined condensation for use as a luminescent ink. Chem. Commun. 2018, 54, 8064–8067. [Google Scholar] [CrossRef]
- Li, Z.-J.; Hofman, E.; Li, J.; Davis, A.H.; Tung, C.-H.; Wu, L.-Z.; Zheng, W. Photoelectrochemically Active and Environmentally Stable CsPbBr3/TiO2 Core/Shell Nanocrystals. Adv. Funct. Mater. 2018, 28, 1704288. [Google Scholar] [CrossRef]
- Li, Z.; Kong, L.; Huang, S.; Li, L. Highly Luminescent and Ultrastable CsPbBr3 Perovskite Quantum Dots Incorporated into a Silica/Alumina Monolith. Angew. Chem. Int. Ed. 2017, 56, 8134–8138. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, Y.; Cao, M.; Hu, H.; Wu, L.; Yu, X.; Wang, L.; Sun, B.; Zhang, Q. Fabricating CsPbX3-Based Type I and Type II Heterostructures by Tuning the Halide Composition of Janus CsPbX3/ZrO2 Nanocrystals. ACS Nano 2019, 13, 5366–5374. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, B.; Wang, Z.; Hung, T.F.; Susha, A.S.; Zhong, H.; Rogach, A.L. Water resistant CsPbX3 nanocrystals coated with polyhedral oligomeric silsesquioxane and their use as solid state luminophores in all-perovskite white light-emitting devices. Chem. Sci. 2016, 7, 5699–5703. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yuan, G.; Lu, Z.; Xia, J.; Li, D.; Chen, Y.; Zhang, Y.; Pei, F.; Chen, C.; Bai, Y.; et al. Engineering Amorphous–Crystallized Interface of ZrNx Barriers for Stable Inverted Perovskite Solar Cells. Adv. Mater. 2023, 35, 2301684. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.N.; Bekenstein, Y.; Koc, M.A.; Fischer, S.; Zhang, D.; Lin, L.; Ritchie, R.O.; Yang, P.; Alivisatos, A.P. Encapsulation of Perovskite Nanocrystals into Macroscale Polymer Matrices: Enhanced Stability and Polarization. ACS Appl. Mater. Interfaces 2016, 8, 35523–35533. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, S.; Cao, F.; Zhou, J.; Wu, Q.; Wang, H.; Li, X.; Yin, L.; Yang, X. Ultrastable Inorganic Perovskite Nanocrystals Coated with a Thick Long-Chain Polymer for Efficient White Light-Emitting Diodes. Chem. Mater. 2019, 31, 1936–1940. [Google Scholar] [CrossRef]
- Pan, A.; Wang, J.; Jurow, M.J.; Jia, M.; Liu, Y.; Wu, Y.; Zhang, Y.; He, L.; Liu, Y. General Strategy for the Preparation of Stable Luminous Nanocomposite Inks Using Chemically Addressable CsPbX3 Peroskite Nanocrystals. Chem. Mater. 2018, 30, 2771–2780. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Mu, Y.-F.; Guo, X.-X.; Zhang, W.; Zhang, Z.-M.; Zhang, M.; Lu, T.-B. Encapsulating Perovskite Quantum Dots in Iron-Based Metal–Organic Frameworks (MOFs) for Efficient Photocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 9491–9495. [Google Scholar] [CrossRef]
- Chen, S.; Yin, H.; Liu, P.; Wang, Y.; Zhao, H. Stabilization and Performance Enhancement Strategies for Halide Perovskite Photocatalysts. Adv. Mater. 2023, 35, 2203836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Guhrenz, C.; Kirch, A.; Sonntag, L.; Bauer, C.; Fan, X.; Wang, J.; Reineke, S.; Gaponik, N.; Eychmüller, A. Highly Luminescent and Water-Resistant CsPbBr3–CsPb2Br5 Perovskite Nanocrystals Coordinated with Partially Hydrolyzed Poly(methyl methacrylate) and Polyethylenimine. ACS Nano 2019, 13, 10386–10396. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lv, Y.; Cao, B.; Wang, W. Surface Modification Strategy Synthesized CsPbX3 Perovskite Quantum Dots with Excellent Stability and Optical Properties in Water. Adv. Funct. Mater. 2023, 33, 2300493. [Google Scholar] [CrossRef]
- Jagadeeswararao, M.; Vashishtha, P.; Hooper, T.J.N.; Kanwat, A.; Lim, J.W.M.; Vishwanath, S.K.; Yantara, N.; Park, T.; Sum, T.C.; Chung, D.S.; et al. One-Pot Synthesis and Structural Evolution of Colloidal Cesium Lead Halide–Lead Sulfide Heterostructure Nanocrystals for Optoelectronic Applications. J. Phys. Chem. Lett. 2021, 12, 9569–9578. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Patra, A.; Dutta, S.K.; Shyamal, S.; Pradhan, N. Facets-Directed Epitaxially Grown Lead Halide Perovskite-Sulfobromide Nanocrystal Heterostructures and Their Improved Photocatalytic Activity. J. Am. Chem. Soc. 2022, 144, 18629–18641. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Patra, A.; Shyamal, S.; Nasipuri, D.; Pradhan, N. Lead Halide Perovskite Cube Coupled Star Nanocrystal Photocatalysts. ACS Energy Lett. 2022, 7, 3015–3023. [Google Scholar] [CrossRef]
- Rusch, P.; Toso, S.; Ivanov, Y.P.; Marras, S.; Divitini, G.; Manna, L. Nanocrystal Heterostructures Based On Halide Perovskites and Lead–Bismuth Chalcogenides. Chem. Mater. 2023, 35, 10684–10693. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, C.; Zhang, P.; Ma, S.; Chen, Y.; Zhang, Y.; Yang, N.; Xiao, M.; Cheng, X.; Gao, Z.; et al. Topochemical assembly minimizes lattice heterogeneity in polycrystalline halide perovskites. Joule 2023, 7, 2361–2375. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, D.; Li, Y.; Xia, J.; Wei, H.; Ding, C.; Hu, Y.; Wei, Y.; Li, H.; Liu, D.; et al. In Situ Room-Temperature Synthesis of All-Colloidal Quantum Dot CsPbBr3–PbS Heterostructures. ACS Photonics 2023, 10, 4305–4314. [Google Scholar] [CrossRef]
- Gaulding, E.A.; Chen, X.; Yang, Y.; Harvey, S.P.; To, B.; Kim, Y.-H.; Beard, M.C.; Sercel, P.C.; Luther, J.M. Embedding PbS Quantum Dots (QDs) in Pb-Halide Perovskite Matrices: QD Surface Chemistry and Antisolvent Effects on QD Dispersion and Confinement Properties. ACS Mater. Lett. 2020, 2, 1464–1472. [Google Scholar] [CrossRef]
- Yan, D.; Liu, M.; Li, Z.; Hou, B. Colloidal quantum dots and metal halide perovskite hybridization for solar cell stability and performance enhancement. J. Mater. Chem. A 2021, 9, 15522–15541. [Google Scholar] [CrossRef]
- Vashishtha, P.; Nutan, G.V.; Griffith, B.E.; Fang, Y.; Giovanni, D.; Jagadeeswararao, M.; Sum, T.C.; Mathews, N.; Mhaisalkar, S.G.; Hanna, J.V.; et al. Cesium Copper Iodide Tailored Nanoplates and Nanorods for Blue, Yellow, and White Emission. Chem. Mater. 2019, 31, 9003–9011. [Google Scholar] [CrossRef]
- Swarnkar, A.; Mir, W.J.; Chakraborty, R.; Jagadeeswararao, M.; Sheikh, T.; Nag, A. Are Chalcogenide Perovskites an Emerging Class of Semiconductors for Optoelectronic Properties and Solar Cell? Chem. Mater. 2019, 31, 565–575. [Google Scholar] [CrossRef]











































| Photocatalyst | Light Source | Band Gap | Synthesis Method | C % | S % | Activity (µmol·g−1·h−1) | Ref |
|---|---|---|---|---|---|---|---|
| FAPbBr3 | 150 W Xe lamp, AM 1.5 G simulated light irradiation | 2.2 | LARP | 0.075 | 81 | 320 | [97] |
| CsPbBr3 | 300 W Xe lamp with 420 nm filter | 2.3 | LARP | - | - | 710 | [124] |
| CdS | 300 W Xe lamp, visible light (λ > 420 nm) | 2.25 | room temp. | 33 | 100 | 240 | [167] |
| TiO2 | Six 6 W UV lamps (310 nm) | ~3.7 | Hydrothermal | 21% | 90 | 94 | [168] |
| Flower-like Bi2WO6 | 300 W Xe lamp, visible light (λ > 420 nm) | 2.96 | Hydrothermal | 1.5% | ~85 | 464 | [169] |
| CsPbBr3−xClx/TiO2 | 300 W Xe lamp with 420 nm filter | 2.62 | LARP & anion exchange | - | - | 1874 | [124] |
| 5% NiOx/FAPbBr3/TiO2 | 150 W Xe lamp, AM 1.5 G | - | LARP | 0.85% | 86 | 3800 | [97] |
| Cs3Bi2Br9 NCs | 300 W Xe lamp | 2.75 | LARP | - | 100 | 850 | [141] |
| Cs3Bi2Br9/Mxene-7.5 | 300 W Xe lamp | ~2.5 | In-situ growth | - | - | 4011 | [141] |
| Cs3Bi2Br9/SBA-15(10%) | 300 W Xe lamp with 420 nm filter | 2.74 | wetness impregnation | - | 90 | 12,600 | [160] |
| Cs3Sb2Br9 | 150 W Xe lamp, AM 1.5 G with 420 nm filter | 2.3 | Anti-solvent precipitation | - | - | 1195 | [148] |
| g-C3N4 | 150 W Xe lamp, AM 1.5 G with 420 nm filter | 2.67 | Pyrolysis | - | - | 298.3 | [148] |
| Cs3Sb2Br9/g-C3N4 | 150 W Xe lamp, AM 1.5 G with 420 nm filter | 2.3 | Anti-solvent precipitation | - | - | 8347 | [148] |
| Cs2AgBiBr6 | 500 W Xe lamp, with 420 nm filter | 2.21 | hydrothermal | - | 10 | 53 | [165] |
| Cs2AgSbxBi1−xBr6 | 500 W Xe lamp, with 420 nm filter | 2.07 | hydrothermal | - | 40 | 448 | [165] |
| Cs2AgSbxBi1−xBr6/CN (20%) | 500 W Xe lamp, with 420 nm filter | - | Mechanical grinding | - | 96 | 1088 | [165] |
| Cs4ZnSb2Cl12 nanoplates | 500 W Xe lamp, with 420 nm filter | 2.36 | Hot injection | - | 95 | 1893 | [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jagadeeswararao, M.; Galian, R.E.; Pérez-Prieto, J. Photocatalysis Based on Metal Halide Perovskites for Organic Chemical Transformations. Nanomaterials 2024, 14, 94. https://doi.org/10.3390/nano14010094
Jagadeeswararao M, Galian RE, Pérez-Prieto J. Photocatalysis Based on Metal Halide Perovskites for Organic Chemical Transformations. Nanomaterials. 2024; 14(1):94. https://doi.org/10.3390/nano14010094
Chicago/Turabian StyleJagadeeswararao, Metikoti, Raquel E. Galian, and Julia Pérez-Prieto. 2024. "Photocatalysis Based on Metal Halide Perovskites for Organic Chemical Transformations" Nanomaterials 14, no. 1: 94. https://doi.org/10.3390/nano14010094