

Exploring the Influence of P3HT on PTCA Crystallization and Phase Behavior in Thin Films
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
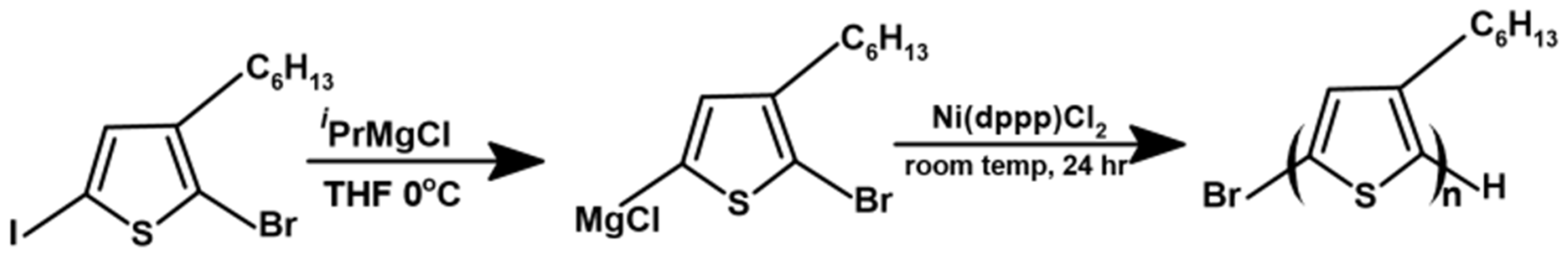
2.2. Synthesis of P3HT
2.3. Synthesis of PTCA-SMs
2.4. Thin-Film Preparation
2.5. Characterization
2.5.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.5.2. Fourier-Transform Infrared (FT-IR) Spectroscopy
2.5.3. Size-Exclusion Chromatography
2.5.4. Spectroscopic and Variable-Temperature Ellipsometries
2.5.5. Differential Scanning Calorimetry (DSC) Analysis
2.5.6. Atomic Force Microscopy (AFM) Analysis
2.5.7. X-ray Diffraction (XRD) Analysis
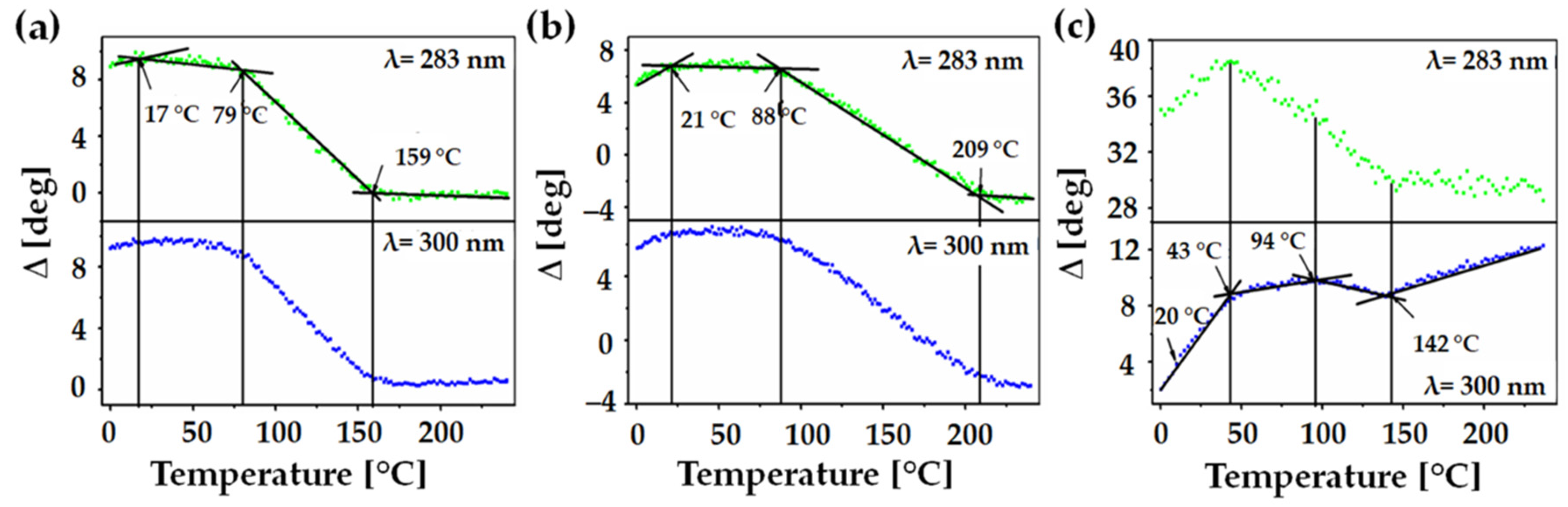
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelley, T.W.; Baude, P.F.; Gerlach, C.; Ender, D.E.; Muyres, D.; Haase, M.A.; Vogel, D.E.; Theiss, S.D. Recent Progress in Organic Electronics: Materials, Devices, and Processes. Chem. Mater. 2004, 16, 4413–4422. [Google Scholar] [CrossRef]
- Friederich, P.; Fediai, A.; Kaiser, S.; Konrad, M.; Jung, N.; Wenzel, W. Toward Design of Novel Materials for Organic Electronics. Adv. Mater. 2019, 31, 1808256. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.N.; Lee, H.; Lee, H.C.; Cho, K. van der Waals Epitaxy of Organic Semiconductor Thin Films on Atomically Thin Graphene Templates for Optoelectronic Applications. Acc. Chem. Res. 2022, 55, 673–684. [Google Scholar] [CrossRef]
- Peet, J.; Heeger, A.J.; Bazan, G.C. The Role of Processing in the Fabrication and Optimization of Plastic Solar Cells. Acc. Chem. Res. 2009, 42, 1700–1708. [Google Scholar] [CrossRef]
- Root, E.S.; Savagatrup, S.; Printz, A.D.; Rodriquez, D.; Lipomi, D.J. Mechanical Properties of Organic Semiconductors for Stretchable, Highly Flexible, and Mechanically Robust Electronics. Chem. Rev. 2017, 117, 6467–6499. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Asare-Yeboah, K.; Zhang, Z.; Bi, S. Manipulate Organic Crystal Morphology and Charge Transport. Org. Electron. 2022, 103, 106448. [Google Scholar] [CrossRef]
- Chen, J.; Shao, M.; Xiao, K.; He, Z.; Li, D.; Lokitz, B.S.; Hensley, D.K.; Kilbey, S.M.; Anthony, J.E.; Keum, J.K.; et al. Conjugated Polymer-Mediated Polymorphism of a High Performance, Small-molecule Organic Semiconductor with Tuned Intermolecular Interactions, Enhanced Long-range Order, and Charge Transport. Chem. Mater. 2013, 25, 4378–4386. [Google Scholar] [CrossRef]
- Bi, S.; Li, Y.; He, Z.; Ouyang, Z.; Guo, Q.; Jiang, C. Self-Assembly Diketopyrrolopyrrole-Based Materials and Polymer Blend with Enhanced Crystal Alignment and Property for Organic Field-Effect Transistor. Org. Electron. 2019, 65, 96–99. [Google Scholar] [CrossRef]
- Zheng, Y.; Xue, J. Organic Photovoltaic Cells Based on Molecular Donor-Acceptor Heterojunctions. Polym. Rev. 2010, 50, 420–453. [Google Scholar] [CrossRef]
- Ng, L.W.T.; Lee, S.W.; Chang, D.W.; Hodgkiss, J.M.; Vak, D. Organic photovoltaics’ New Renaissance: Advances toward Roll-to-roll Manufacturing of Non-Fullerene Acceptor Organic Photovoltaics. Adv. Mater. Technol. 2022, 7, 2101556. [Google Scholar] [CrossRef]
- Chen, H. The Effect of Ring Expansion in Thienobenzo[b]indacenodithiophene Polymers for Organic Field-Effect Transistors. J. Am. Chem. Soc. 2019, 141, 18806–18813. [Google Scholar] [CrossRef] [PubMed]
- Gunes, S.; Neugebauer, H.; Sariciftci, N.S. Conjugated Polymer-Based Organic Solar Cells. Chem. Rev. 2007, 107, 1324–1338. [Google Scholar] [CrossRef]
- Ibanez, J.G.; Rincon, M.E.; Gutierrez-Granados, S. Conducting Polymers in the Fields of Energy, Environmental Remediation, and Chemical-chiral Sensors. Chem. Rev. 2018, 118, 4731–4816. [Google Scholar] [CrossRef] [PubMed]
- Khawas, K.; Daripa, S.; Kumari, P.; Das, S.; Dey, R.; Kuila, B. Highly Water-Soluble Rod–Coil Conjugated Block Copolymer for Efficient Humidity Sensor. Macro. Molecul. Chem. Phy. 2019, 220, 1900013. [Google Scholar] [CrossRef]
- Jadoun, S.; Riaz, U. Conjugated Polymer Light-Emitting Diodes. In Polymers for Light-Emitting Devices and Displays; Scrivener Publishing LLC: Beverly, MA, USA, 2020; pp. 77–98. [Google Scholar]
- Kim, J.Y.; Lee, K.; Coates, N.E.; Moses, D.; Nguyen, T.-Q.; Dante, M.; Heeger, A.J. Efficient Tandem Polymer Solar Cells Fabricated by all-solution Processing. Science 2007, 317, 222–225. [Google Scholar] [CrossRef]
- Kumari, P.; Bera, M.K.; Malik, S.; Kuila, B.K. Amphiphilic and Thermoresponsive Conjugated Block Copolymer with Its Solvent Dependent Optical and Photoluminescence Properties: Toward Sensing Applications. ACS Appl. Mater. Interfaces 2015, 7, 12348–12354. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, Z.; Bi, S.; Chen, J.; Li, D. Conjugated Polymer Controlled Morphology and Charge Transport of Small-Molecule Organic Semiconductors. Sci. Rep. 2020, 10, 4344. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, G.; Kouki, F.; Spearman, P.; Fichou, D.; Nogues, C.; Pan, X.; Garnier, F. Evidence for n-type Conduction in a Perylene Tetracarboxylic Diimide Derivative. Adv. Mater. 1996, 8, 242–245. [Google Scholar] [CrossRef]
- Jancy, B.; Asha, S.K. Control of Molecular Structure in the Generation of Highly Luminescent Liquid Crystalline Perylenebisimide Derivatives: Synthesis, Liquid Crystalline and Photophysical Properties. J. Phys. Chem. B 2006, 110, 20937–20947. [Google Scholar] [CrossRef]
- Liang, Q.; Lu, H.; Chang, Y.; He, Z.; Zhao, Y.; Liu, J. Morphology Control in Organic Solar Cells. Energies 2022, 15, 5344. [Google Scholar] [CrossRef]
- Ajay, V.; Stefan, M.; Zhenan, B.; Natalie, S. Organic Semiconductor Growth and Morphology Considerations for Organic Thin-Film Transistors. Adv. Mater. 2010, 22, 3857–3875. [Google Scholar] [CrossRef]
- Wang, T.; Pearson, A.J.; Dunbar, A.D.F.; Staniec, P.A.; Watters, D.C.; Coles, D.; Yi, H.; Iraqi, A.; Lidzey, D.G.; Jones, R.A.L. Competition Between Substrate-mediated π-π Stacking and Surface Mediated Tg Depression in Ultrathin Conjugated Polymer Films. Eur. Phys. J. E 2012, 35, 129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, J.; Zin, W.-C. Estimation of the Thickness Dependence of the Glass Transition Temperature in Various Thin Polymer Films. Langmuir 2000, 16, 4064–4067. [Google Scholar] [CrossRef]
- Chandran, S.; Basu, J.K. Effect of Nanoparticle Dispersion on Glass Transition in Thin Films of Polymer NanoBlends. Eur. Phys. J. E 2011, 34, 99. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Bergqvist, J.; Vandewal, K.; Tvingstedt, K.; Anselmo, A.S.; Magnusson, R.; Alonso, M.I.; Moons, E.; Arwin, H.; Campoy-Quiles, M.; et al. Phase Behaviour of Liquid-Crystalline Polymer/fullerene Organic Photovoltaic Blends: Thermal Stability and Miscibility. J. Mater. Chem. 2011, 21, 10676–10684. [Google Scholar] [CrossRef]
- Müller, C. On the Glass Transition of Polymer Semiconductors and Its Impact on Polymer Solar Cell Stability. Chem. Mater. 2015, 27, 2740–2754. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, J.; Zin, W.-C. Thickness Dependence of the Glass Transition Temperature in Thin Polymer Films. Langmuir 2001, 17, 2703–2710. [Google Scholar] [CrossRef]
- Sharp, J.S.; Forrest, J.A. Dielectric and Ellipsometric Studies of the Dynamics in thin Films of Isotactic Poly(methylmethacrylate) with one Free Surface. Phys. Rev. E 2003, 67, 031805. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Hewlett, S.A.; Roth, C.B.; Torkelson, J.M. Confinement Effects on Glass Transition Temperature, Transition Breadth, and Expansivity: Comparison of Ellipsometry and Fluorescence Measurements on Polystyrene Films. Eur. Phys. J. E 2009, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Beaucage, C.; Composto, R.; Stein, R.S. Ellipsometric Study of the Glass Transition and Thermal Expansion Coefficients of Thin Polymer Films. J. Polym. Sci. Part B Polym. Phys. 1993, 31, 319–326. [Google Scholar] [CrossRef]
- Jarząbek, B.; Hajduk, B.; Jurusik, J.; Domanski, M. In Situ Optical Studies of Thermal Stability of Iodine-doped Polyazomethine Thin Films. Polym. Test. 2017, 59, 230–236. [Google Scholar] [CrossRef]
- Yokoyama, A.; Miyakoshi, R.; Yokozawa, T. Chain-Growth Polymerization for Poly(3-hexylthiophene) with a Defined Molecular Weight and a Low Polydispersity. Macromolecules 2004, 37, 1169–1171. [Google Scholar] [CrossRef]
- Ferrere, S.; Zaban, A.; Gregg, B.A. Dye Sensitization of Nanocrystalline Tin Oxide by Perylene Derivatives. J. Phys. Chem. B 1997, 101, 4490. [Google Scholar] [CrossRef]
- Sayyad, A.S.; Balakrishnan, K.; Ajayan, P.M. Chemical Reaction Mediated Self-Assembly of PTCDA into Nanofibers. Nanoscale 2011, 3, 3605. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, B.; Bednarski, H.; Jarząbek, B.; Janeczek, H.; Nitschke, P. P3HT:PCBM Blend Films Phase Diagram on the base of Variable-temperature Spectroscopic Ellipsometry. Beilstein J. Nanotechnol. 2018, 9, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, B.; Bednarski, H.; Jarzabek, B.; Nitschke, P.; Janeczek, H. Phase Diagram of P3HT:PC70BM Thin Films Based on Variable-temperature Spectroscopic Ellipsometry. Polym. Test. 2020, 84, 106383. [Google Scholar] [CrossRef]
- Hajduk, B.; Jarka, P.; Tański, T.; Bednarski, H.; Janeczek, H.; Gnida, P.; Fijalkowski, M. An Investigation of the Thermal Transitions and Physical Properties of Semiconducting PDPP4T:PDBPyBT Blend Films. Materials 2022, 15, 8392. [Google Scholar] [CrossRef] [PubMed]
- Swinehart, D.F. The Beer-Lambert Law. J. Chem. Educ. 1962, 39, 333. [Google Scholar] [CrossRef]
- Banerji, N.; Cowan, S.; Vauthey, E.; Heeger, A.J. Ultrafast Relaxation of the Poly(3-hexylthiophene) Emission Spectrum. J. Phys. Chem. C 2011, 115, 9726–9739. [Google Scholar] [CrossRef]
- Talite, M.J.; Huang, H.; Chen, Z.; Li, W.; Cai, K.; Sevilla, R.C.; Soebroto, R.J.; Lin, S.; Chou, W.; Yuan, C. Perylene Tetracarboxylic Acid Crosslinked to Silica Matrix that Enables Ultrahigh Solid-State Quantum Yield and Efficient Photon Recycling for Holographic Luminescent Solar Concentrators. Solar RRL 2022, 6, 2100955. [Google Scholar] [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical Properties and Electronic Structure of Amorphous Germanium. Phys. Status Solidi B 1996, 15, 627–637. [Google Scholar] [CrossRef]
- Amin, P.O.; Ketuly, K.A.; Saeed, S.R.; Muhammadsharif, F.F.; Symes, M.D.; Paul, A.; Sulaiman, K. Synthesis, Spectroscopic, Electrochemical and Photophysical Properties of High Band Gap Polymers for Potential Applications in Semi-Transparent Solar Cells. BMC Chem. 2021, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Jarka, P.; Hajduk, B.; Bednarski, H.; Tański, T. The Investigation of Thermal and Optical Properties of Semiconducting Nanostructural Hybrid Films. Acta Phys. Pol. A 2022, 142, 107–110. [Google Scholar] [CrossRef]
- Campoy-Quiles, M.; Etchegoin, P.G.; Bradley, D.D.C. Exploring the Potential of Ellipsometry for the Characterisation of Electronic, Optical, Morphologic and Thermodynamic Properties of Polyfluorene Thin Films. Synth. Met. 2005, 155, 279–282. [Google Scholar] [CrossRef]
- Hajduk, B.; Bednarski, H.; Domański, M.; Jarząbek, B.; Trzebicka, B. Thermal Transitions in P3HT:PC60BM Films Based on Electrical Resistance Measurements. Polymers 2020, 12, 1458. [Google Scholar] [CrossRef]
- Hajduk, B.; Bednarski, H.; Trzebicka, B. Temperature-Dependent Spectroscopic Ellipsometry of Thin Polymer Films. J. Phys. Chem. B 2020, 124, 3229–3251. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, H.; Hajduk, B.; Domański, M.; Jarząbek, B.; Nitschke, P.; Łaba, K.; Wanic, A.; Łapkowski, M. Unveiling of Polymer/fullerene Blend Films Morphology by Ellipsometrically Determined Optical Order within Polymer and Fullerene Phases. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 1094–1100. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Brown, P.; Friend, R.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.M.W.; Spiering, A.J.H.; Janssen, R.A.J.; Meijer, E.W.; Herwig, P.; et al. Two-dimensional Charge Transport in Self-Organized, High-mobility Conjugated Polymers. Nature 1999, 401, 685–688. [Google Scholar] [CrossRef]
- Richter, U. SpectraRay/3 Software Manual, Sentech Instruments GmbH. 2011. Available online: https://minateh.ru/assets/files/SENTECH/V2012-PD-SpectraRay-3.pdf (accessed on 1 December 2022).
- Raimo, M. Impact of Thermal Properties on Crystalline Structure, Polymorphism and Morphology of Polymer Matrices in Composites. Materials 2021, 14, 2136. [Google Scholar] [CrossRef]
- Nguyen, A.N.; Remy, A.R.; Mackay, M.E. Thermal Analysis of Semiconducting Polymer Crystals Free of a Mobile Amorphous Fraction. Macromolecules 2021, 54, 2155–2161. [Google Scholar] [CrossRef]
- Zhao, A.; Swinnen, G.; Van Assche, J.; Manca, D.; Vanderzande, B.; Van, M. Phase Diagram of P3HT/PCBM Blends and its Implication for the Stability of Morphology. J. Phys. Chem. B 2009, 113, 1587–1591. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.J.; Wang, T.; Jones, R.A.L.; Lidzey, D.G.; Staniec, P.A.; Hopkinson, P.E.; Donald, A.M. Rationalizing Phase Transitions with Thermal Annealing Temperatures for P3HT:PCBM Organic Photovoltaic Devices. Macromolecules 2012, 45, 1499–1508. [Google Scholar] [CrossRef]
- Sivamurugan, V.; Ramunas, L.; Gra, C.; Juozas, G.; Valentas, G.; Jankauskas, V.; Suresh, V. Effect of Substituents on the Electron Transport Properties of Bay Substituted Perylene Diimide Derivatives. J. Mater. Chem. 2009, 19, 4268. [Google Scholar] [CrossRef]
- Zhao, Y.; Yuan, G.; Roche, P.; Leclerc, M.A. Calorimetric Study of the Phase Transitions in Poly(3-hexylthiophene). Polymer 1995, 36, 2211–2214. [Google Scholar] [CrossRef]
- Son, S.Y.; Park, T.; You, W. Understanding of Face-on Crystallites Transitioning to Edge-on Crystallites in Thiophene-Based Conjugated Polymers. Chem. Mater. 2021, 33, 4541–4550. [Google Scholar] [CrossRef]
- Briseno, A.L.; Mannsfeld, S.C.B.; Reese, C.; Hancock, J.M.; Xiong, Y.; Janekhe, S.A.; Bao, Z.; Xia, Y. Perylenediimide Nanowires and Their Use in Fabricating Field-effect Transisitors and Complementary Inverters. Nano Lett. 2007, 7, 2847–2853. [Google Scholar] [CrossRef]
- Bu, L.; Pentzer, E.; Bokel, F.A.; Emrick, T.; Hayward, R.C. Growth of Polythiophene/perylene Tetracarboxydiimide Donor/Acceptor Shish-kebab Nanostructures by Couples Crystal Modification. ACS Nano 2012, 6, 10924–10929. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, Q.; Nan, J.; Shi, W.; Cui, F.; Zhu, Y. Perylenetetracarboxylic Acid Nanosheets with Internal Electric Fields and Anisotropic Charge Migration for Photocatalytic Hydrogen Evolution. Nat. Commun. 2022, 13, 2067. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Stewart Computational Chemistry; MOPAC: Colorado Springs, CO, USA, 2016. [Google Scholar]
- Altomare, A.; Camalli, M.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R. EXPO2009: Structural Solution Based on Powder Data in Direct and Inverse Space. J. Appl. Crystal. 2009, 42, 1197–1202. [Google Scholar] [CrossRef]
- Rezac, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2011, 8, 141–151. [Google Scholar] [CrossRef]
- Ectors, D.; Goetz-Neunhoeffer, F.; Neubauer, J. Domain size anisotropy in the double-Voigt approach: An extended model. J. Appl. Crystallogr. 2015, 48, 1998–2001. [Google Scholar] [CrossRef]
- Abdou, M.S.A.; Lu, X.; Xie, Z.W.; Orfino, F.; Deen, M.J.; Holdcroft, S. Nature of Impurities in .pi.-Conjugated Polymers Prepared by Ferric Chloride and Their Effect on the Electrical Properties of Metal-Insulator-Semiconductor Structures. Chem. Mater. 1995, 7, 631. [Google Scholar] [CrossRef]
- Grecu, S.; Roggenbuck, M.; Opitz, A.; Brütting, W. Differences of Interface and Bulk Transport Properties in Polymer Field-Effect Devices. Org. Electron. 2006, 7, 276–286. [Google Scholar] [CrossRef]
- Nguyen, N.A.; Shen, H.; Liu, Y.; Mackay, M.E. Kinetics and Mechanism of Poly(3-hexylthiophene) Crystallization in Solution under Shear Flow. Macromolecules 2020, 53, 5795–5804. [Google Scholar] [CrossRef]
- He, Z.; Lopez, N.; Chi, X.; Li, D. Solution-Based 5,6,11,12-Tetrachlorotetracene Crystal Growth for High-Performance Organic Thin Film Transistors. Org. Electron. 2015, 22, 191–196. [Google Scholar] [CrossRef]
- He, Z.; Asare-Yeboah, K.; Zhang, Z.; Bi, S. Self-assembly Crystal Microribbons with Nucleation Additive for High-Performance Organic Thin Film Transistors. Jpn. J. Appl. Phys. 2019, 58, 061009. [Google Scholar] [CrossRef]
- Borgesi, A.; Tallarida, G.; Amore, G.; Cazzaniga, F.; Queirolo, F.; Alessandri, M.; Sassela, A. Influence of roughness and grain dimension on the optical functions of polycrystalline silicon films. Thin Solid Film 1998, 313–314, 243–247. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, P.; Hajduk, B.; Bednarski, H.; Jarka, P.; Janeczek, H.; Łapkowski, M. Exploring the Influence of P3HT on PTCA Crystallization and Phase Behavior in Thin Films. Nanomaterials 2023, 13, 2918. https://doi.org/10.3390/nano13222918
Kumari P, Hajduk B, Bednarski H, Jarka P, Janeczek H, Łapkowski M. Exploring the Influence of P3HT on PTCA Crystallization and Phase Behavior in Thin Films. Nanomaterials. 2023; 13(22):2918. https://doi.org/10.3390/nano13222918
Chicago/Turabian StyleKumari, Pallavi, Barbara Hajduk, Henryk Bednarski, Paweł Jarka, Henryk Janeczek, and Mieczysław Łapkowski. 2023. "Exploring the Influence of P3HT on PTCA Crystallization and Phase Behavior in Thin Films" Nanomaterials 13, no. 22: 2918. https://doi.org/10.3390/nano13222918