
Exchange Bias Demonstrated in Bulk Nanocomposites Processed by High-Pressure Torsion
 , , and
, , and
Abstract
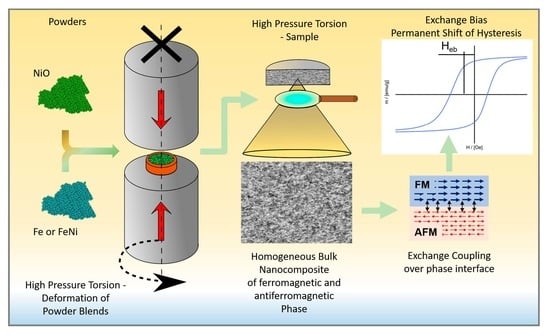
:
1. Introduction
2. Materials and Methods
3. Results
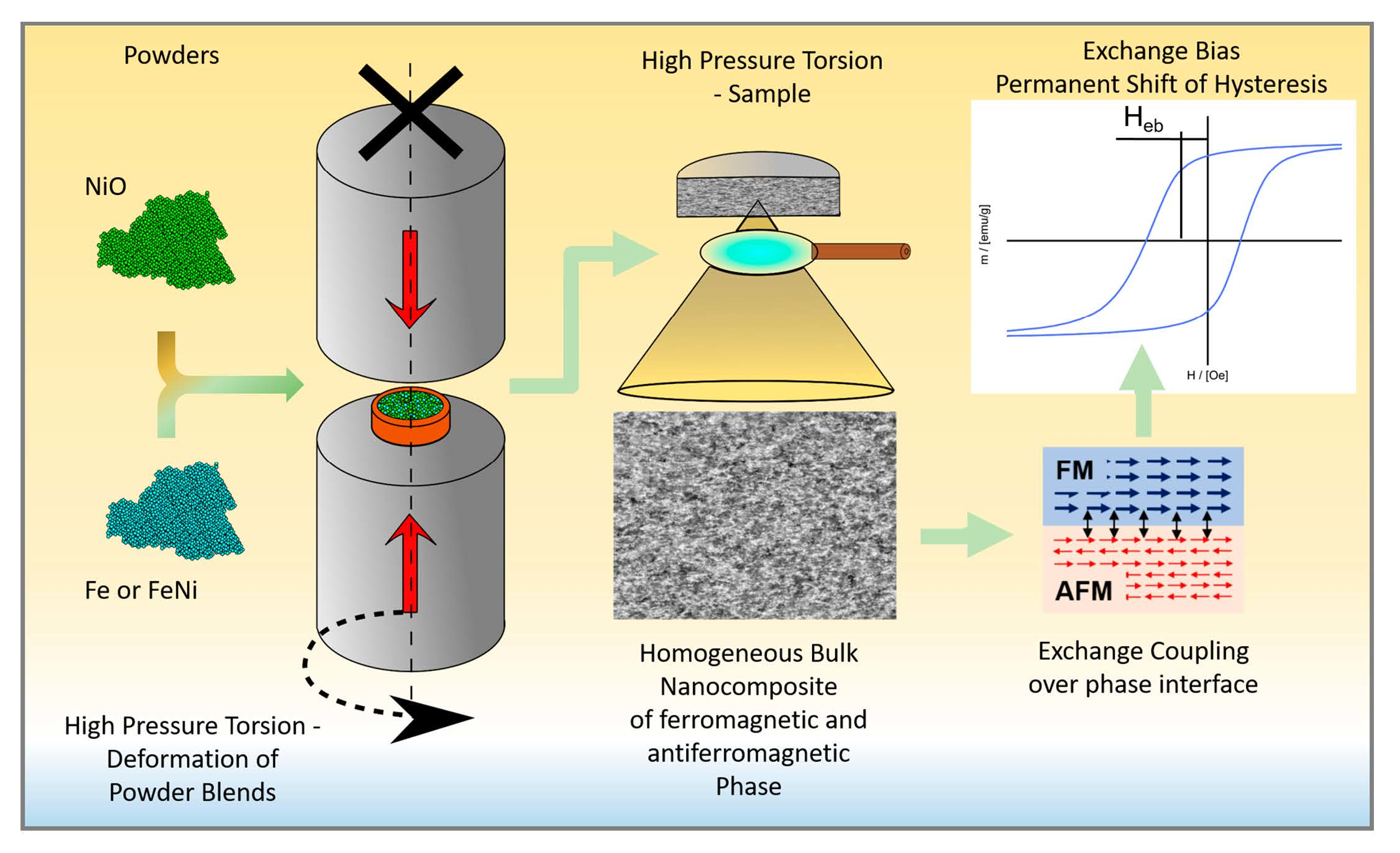
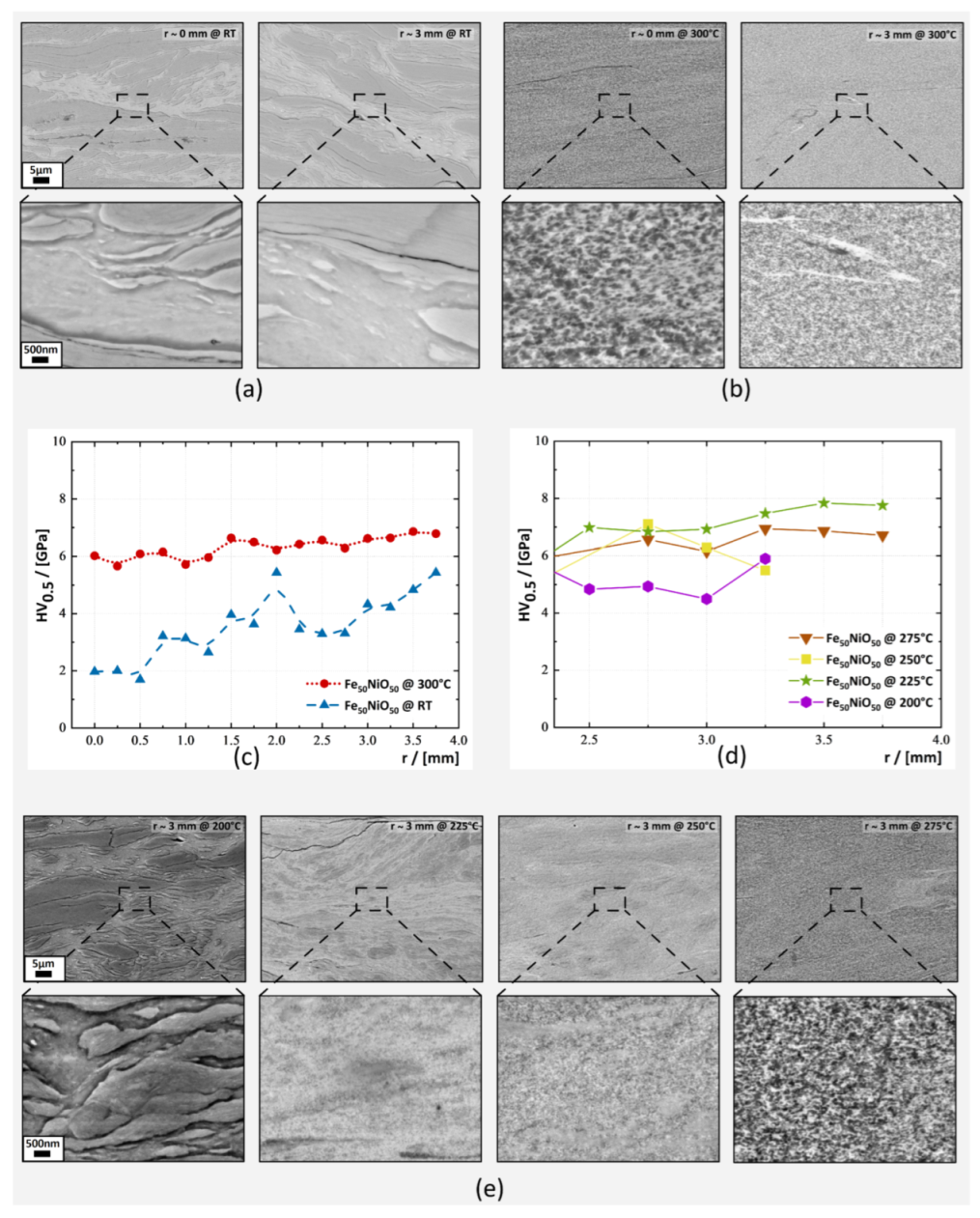
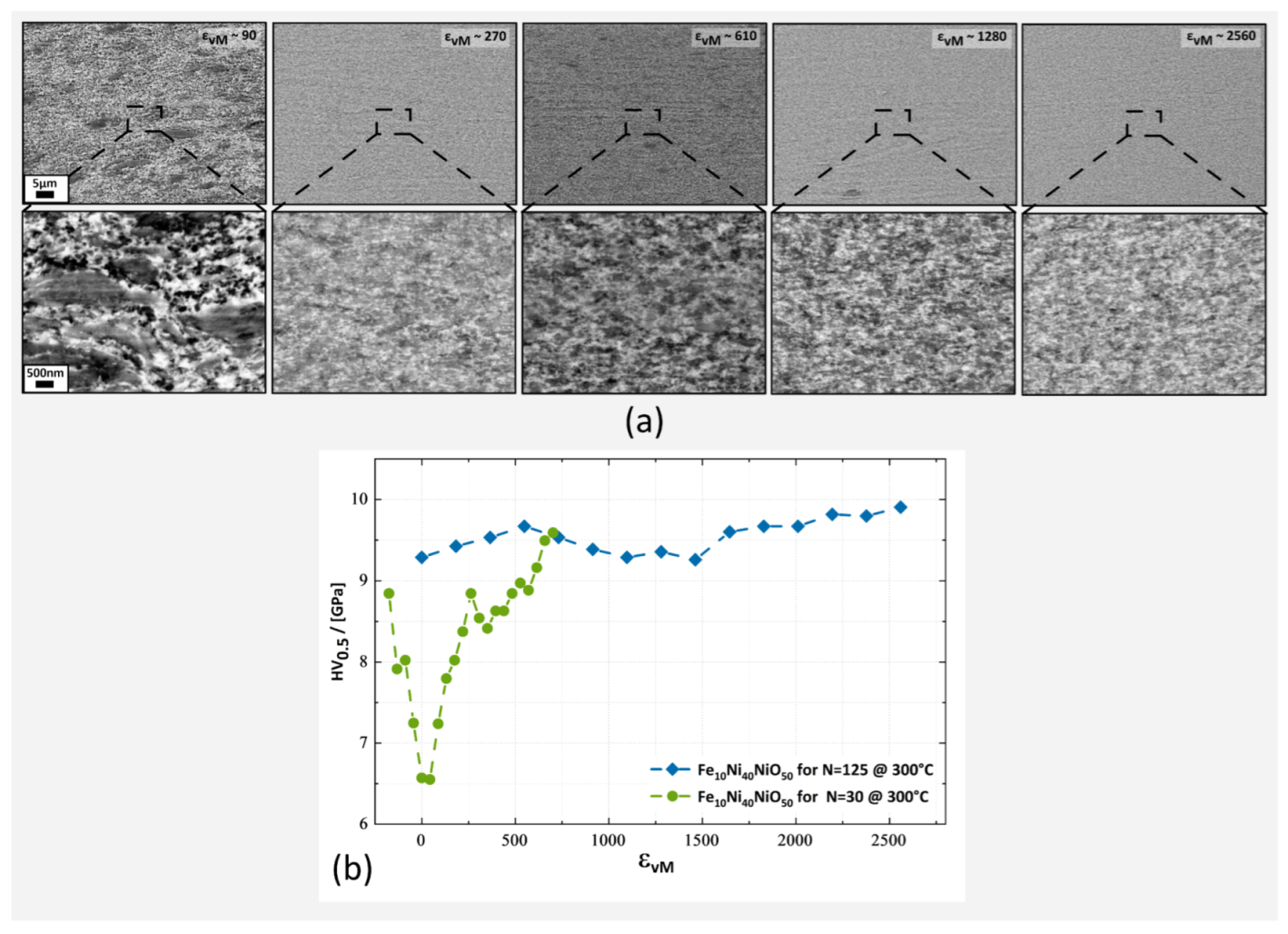
3.1. Hardness and Microstructural Evolution
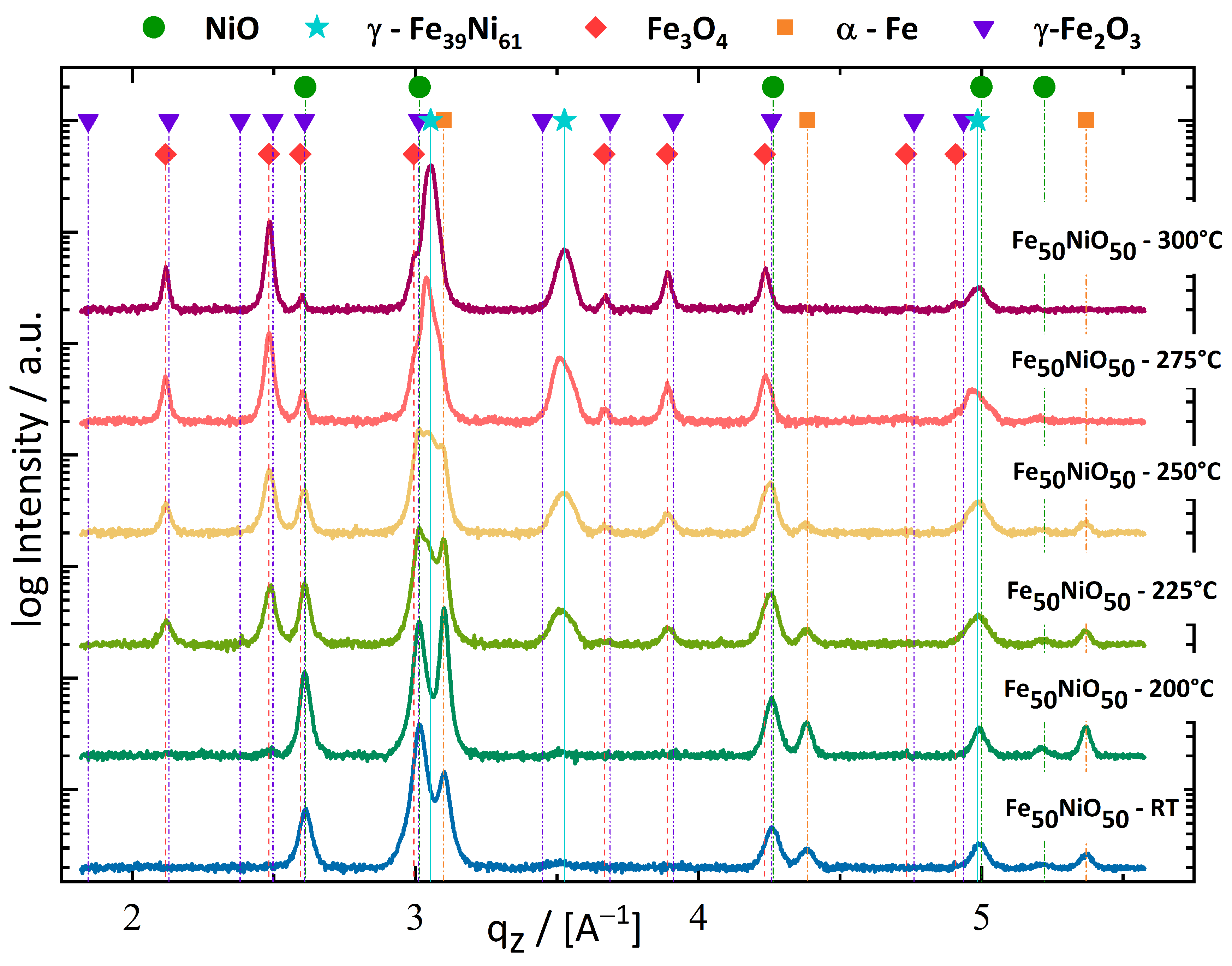
3.1.1. Fe50NiO50 Composition
3.1.2. Fe10Ni40NiO50 Composition
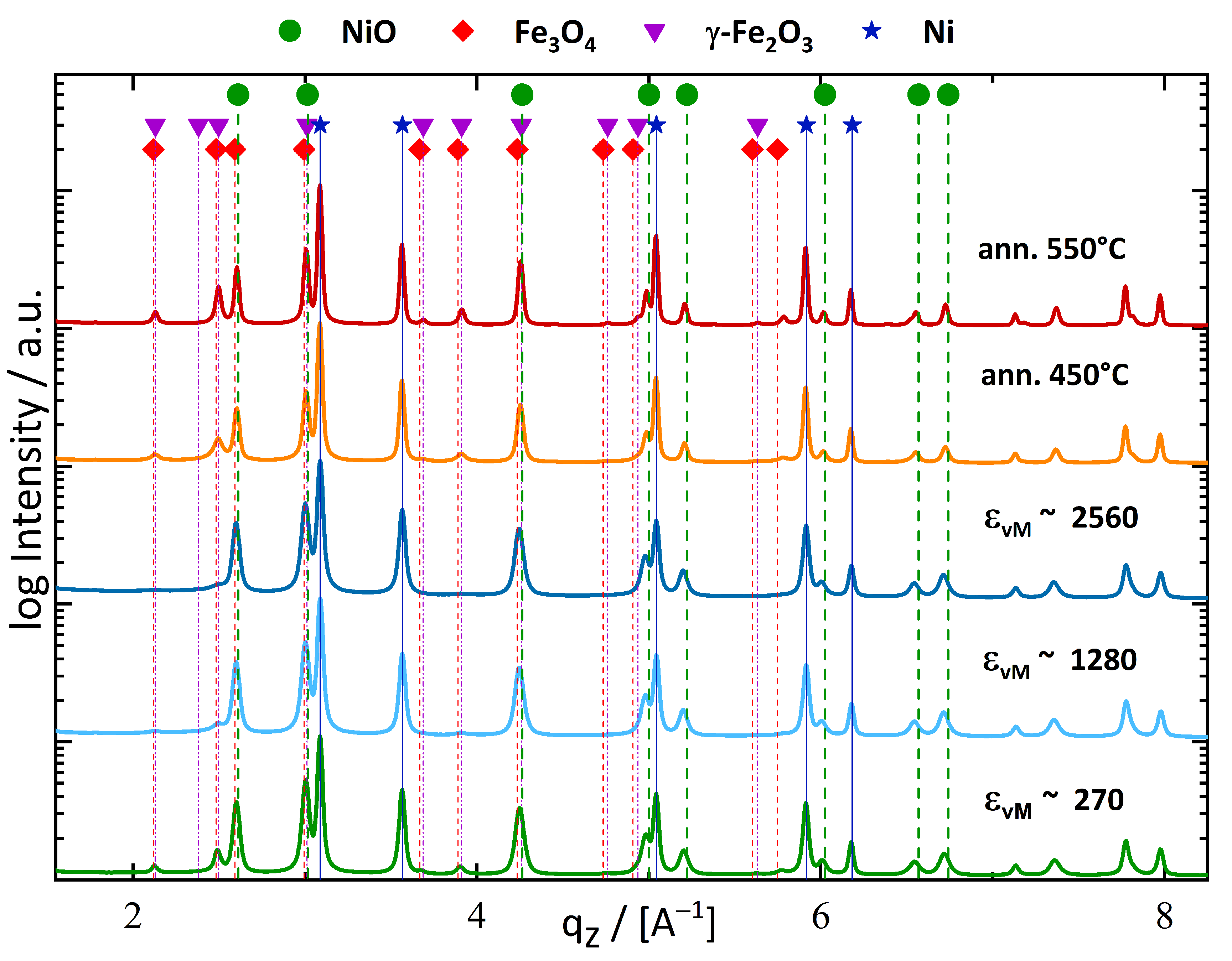
3.1.3. Annealing of Fe10Ni40NiO50 Composition
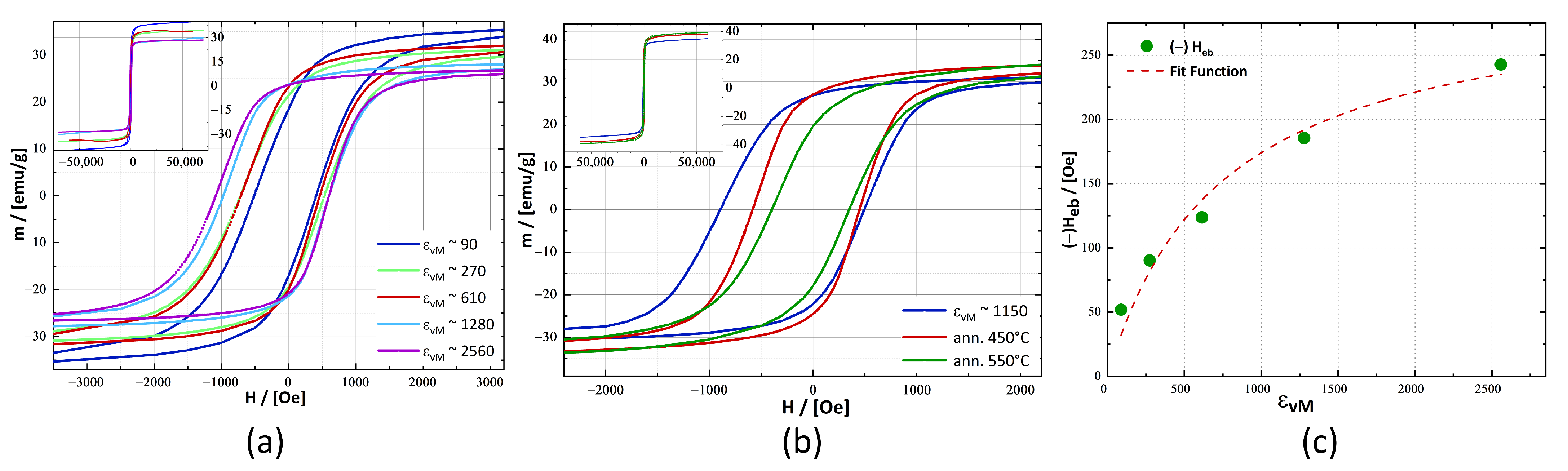
3.2. Magnetic Characterisation
4. Discussion
4.1. Deformation Behaviour
4.2. Phase Formation during Deformation
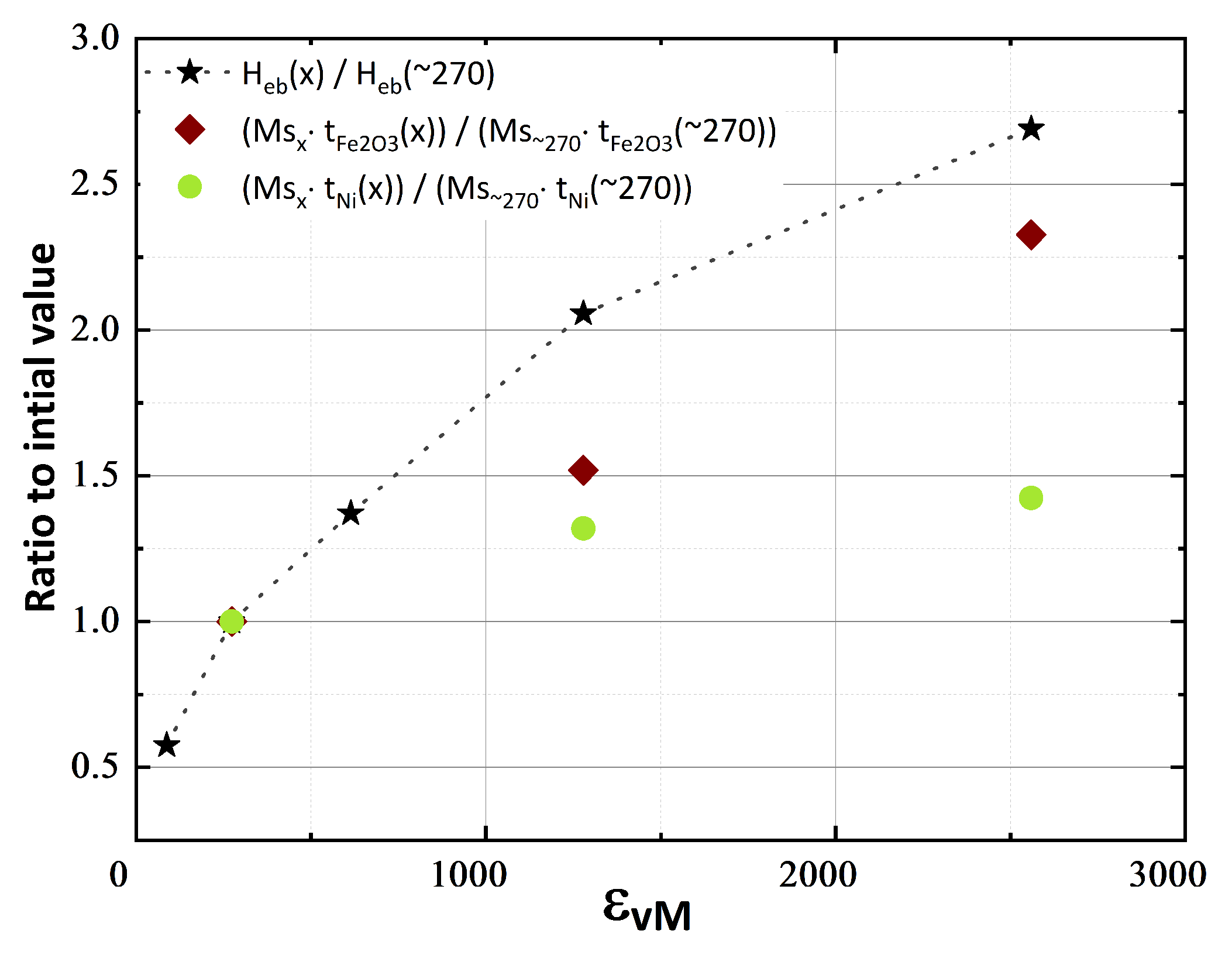
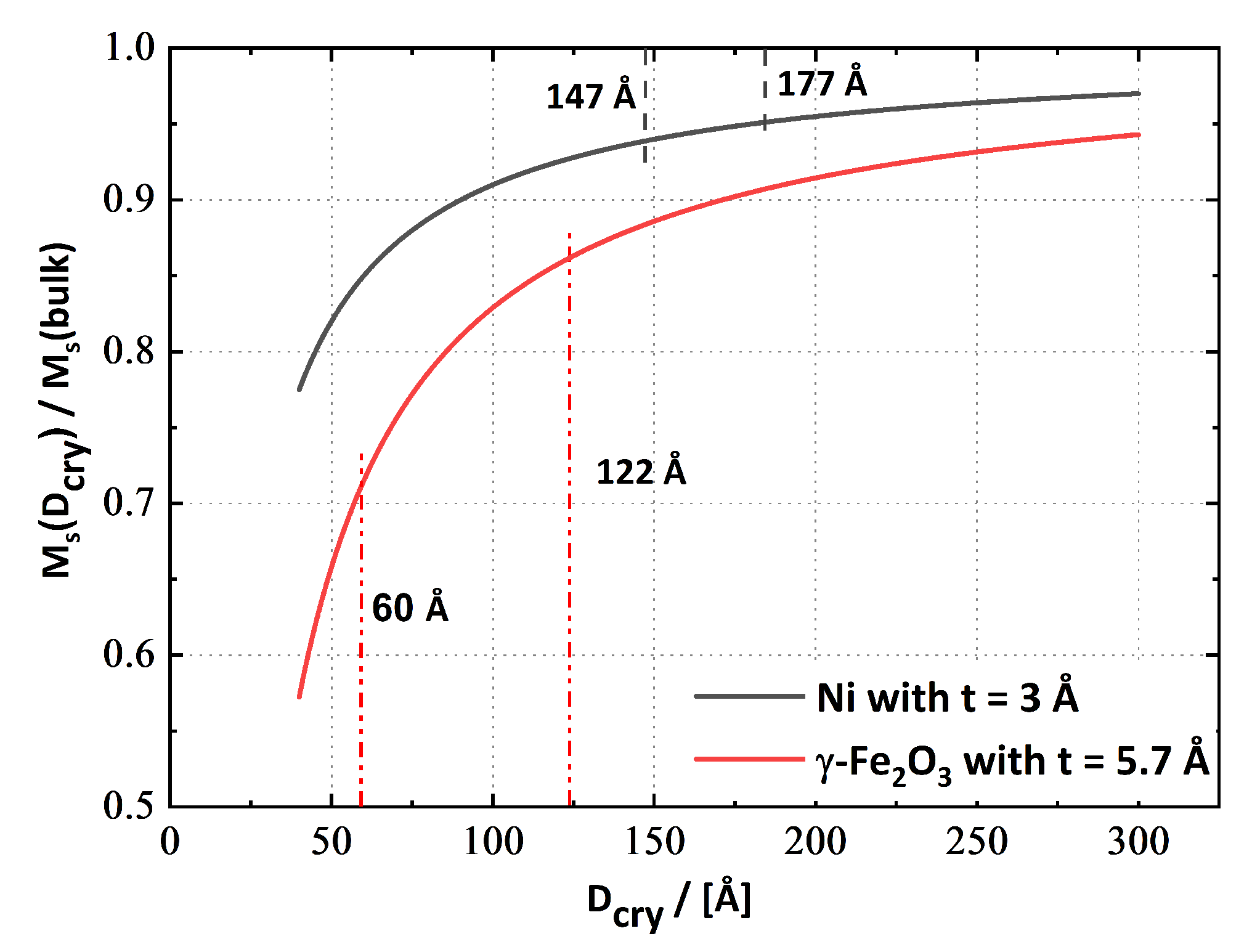
4.3. Magnetic Properties
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
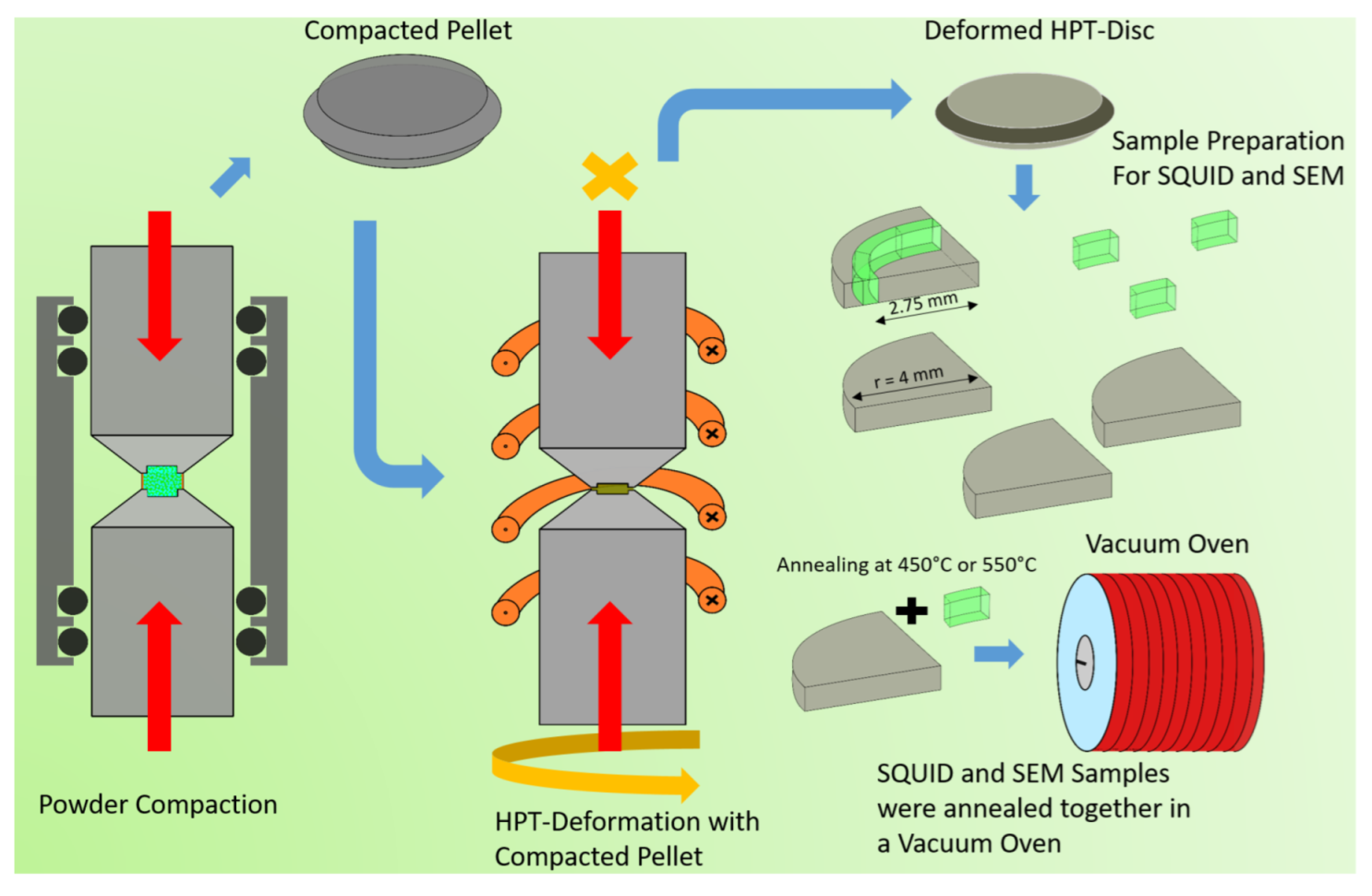
Appendix A. Schematic Illustration of the HPT-Synthesis

References
- Sahu, T.; Ratre, Y.K.; Chauhan, S.; Bhaskar, L.V.; Nair, M.P.; Verma, H.K. Nanotechnology based drug delivery system: Current strategies and emerging therapeutic potential for medical science. J. Drug Deliv. Sci. Technol. 2021, 63, 102487. [Google Scholar] [CrossRef]
- Mitchell, S.; Qin, R.; Zheng, N.; Pérez-Ramírez, J. Nanoscale engineering of catalytic materials for sustainable technologies. Nat. Nanotechnol. 2021, 16, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Z.; Zhao, S.; Xiang, S.; Gao, W.; Wang, L.; Xu, J.; Wang, Y. The promoting effect of alkali metal and H2O on Mn-MOF derivatives for toluene oxidation: A combined experimental and theoretical investigation. J. Catal. 2022, 415, 218–235. [Google Scholar] [CrossRef]
- Nikolic, M.V.; Milovanovic, V.; Vasiljevic, Z.Z.; Stamenkovic, Z. Semiconductor gas sensors: Materials, technology, design, and application. Sensors 2020, 20, 6694. [Google Scholar] [CrossRef]
- Sreenivasulu, V.B.; Narendar, V. A Comprehensive Analysis of Junctionless Tri-Gate (TG) FinFET Towards Low-Power and High-Frequency Applications at 5-nm Gate Length. Silicon 2022, 14, 2009–2021. [Google Scholar] [CrossRef]
- Ovid’ko, I.A.; Valiev, R.Z.; Zhu, Y.T. Review on superior strength and enhanced ductility of metallic nanomaterials. Prog. Mater. Sci. 2018, 94, 462–540. [Google Scholar] [CrossRef]
- Valiev, R.Z.; Estrin, Y.; Horita, Z.; Langdon, T.G.; Zehetbauer, M.J.; Zhu, Y.T. Fundamentals of superior properties in bulk NanoSPD materials. Mater. Res. Lett. 2016, 4, 1–21. [Google Scholar] [CrossRef]
- Herzer, G. Modern soft magnets: Amorphous and nanocrystalline materials. Acta Mater. 2013, 61, 718–734. [Google Scholar] [CrossRef]
- Coey, J.M. Hard magnetic materials: A perspective. IEEE Trans. Magn. 2011, 47, 4671–4681. [Google Scholar] [CrossRef]
- Skomski, R.; Coey, J.M. Magnetic anisotropy-How much is enough for a permanent magnet? Scr. Mater. 2016, 112, 3–8. [Google Scholar] [CrossRef]
- Gutfleisch, O.; Willard, M.A.; Brück, E.; Chen, C.H.; Sankar, S.G.; Liu, J.P. Magnetic materials and devices for the 21st century: Stronger, lighter, and more energy efficient. Adv. Mater. 2011, 23, 821–842. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.A.; Mishra, A.; Benson, D.J. Mechanical properties of nanocrystalline materials. Prog. Mater. Sci. 2006, 51, 427–556. [Google Scholar] [CrossRef]
- Kormout, K.S.; Pippan, R.; Bachmaier, A. Deformation-Induced Supersaturation in Immiscible Material Systems during High-Pressure Torsion. Adv. Eng. Mater. 2017, 19, 1600675. [Google Scholar] [CrossRef]
- Wurster, S.; Weissitsch, L.; Stückler, M.; Knoll, P.; Krenn, H.; Pippan, R.; Bachmaier, A. Tuneable magneto-resistance by severe plastic deformation. Metals 2019, 9, 1188. [Google Scholar] [CrossRef] [Green Version]
- Bachmaier, A.; Krenn, H.; Knoll, P.; Aboulfadl, H.; Pippan, R. Tailoring the magnetic properties of nanocrystalline Cu-Co alloys prepared by high-pressure torsion and isothermal annealing. J. Alloys Compd. 2017, 725, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Weissitsch, L.; Stückler, M.; Wurster, S.; Knoll, P.; Krenn, H.; Pippan, R.; Bachmaier, A. Strain induced anisotropic magnetic behaviour and exchange coupling effect in Fe-SmCo5 permanent magnets generated by high pressure torsion. Crystals 2020, 10, 1026. [Google Scholar] [CrossRef]
- Menéndez, E.; Sort, J.; Langlais, V.; Zhilyaev, A.; Muñoz, J.S.; Suriñach, S.; Nogués, J.; Baró, M.D. Cold compaction of metal-ceramic (ferromagnetic-antiferromagnetic) composites using high pressure torsion. J. Alloys Compd. 2007, 434–435, 505–508. [Google Scholar] [CrossRef]
- Pippan, R.; Wetscher, F.; Hafok, M.; Vorhauer, A.; Sabirov, I. The limits of refinement by severe plastic deformation. Adv. Eng. Mater. 2006, 8, 1046–1056. [Google Scholar] [CrossRef]
- Meiklejohn, W.H.; Bean, C.P. New magnetic anisotropy. Phys. Rev. 1956, 102, 1413–1414. [Google Scholar] [CrossRef]
- Blachowicz, T.; Ehrmann, A. Exchange bias in thin films—an update. Coatings 2021, 11, 122. [Google Scholar] [CrossRef]
- Childress, J.R.; Fontana, R.E. Magnetic recording read head sensor technology. Comptes Rendus Phys. 2005, 6, 997–1012. [Google Scholar] [CrossRef]
- Edalati, K. Review on Recent Advancements in Severe Plastic Deformation of Oxides by High-Pressure Torsion (HPT). Adv. Eng. Mater. 2019, 21, 1800272. [Google Scholar] [CrossRef]
- Hohenwarter, A.; Bachmaier, A.; Gludovatz, B.; Scheriau, S.; Pippan, R. Technical parameters affecting grain refinement by high pressure torsion. Int. J. Mater. Res. 2009, 100, 1653–1661. [Google Scholar] [CrossRef]
- Valiev, R.; Islamgaliev, R.; Alexandrov, I. Bulk nanostructured materials from severe plastic deformation. Prog. Mater. Sci. 2000, 45, 103–189. [Google Scholar] [CrossRef]
- Srinivasan, G.; Seehra, M.S. Magnetic susceptibilities, their temperature variation, and exchange constants of NiO. Phys. Rev. B 1984, 29, 6295–6298. [Google Scholar] [CrossRef]
- Owen, E.A.; Yates, E.L.; Sully, A.H. An X-ray investigation of pure iron-nickel alloys. Part 4: The variation of lattice-parameter with composition. Proc. Phys. Soc. 1937, 49, 315–322. [Google Scholar] [CrossRef]
- Siow, K.S.; Tay, A.A.; Oruganti, P. Mechanical properties of nanocrystalline copper and nickel. Mater. Sci. Technol. 2004, 20, 285–294. [Google Scholar] [CrossRef]
- Bachmaier, A.; Hohenwarter, A.; Pippan, R. New procedure to generate stable nanocrystallites by severe plastic deformation. Scr. Mater. 2009, 61, 1016–1019. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, S.; Gupta, A. Effect of interface roughness on exchange coupling in polycrystalline Co/CoO bilayer structure: An in-situ investigation. J. Appl. Phys. 2016, 120, 085307. [Google Scholar] [CrossRef]
- Nogués, J.; Moran, T.J.; Lederman, D.; Schuller, I.K.; Rao, K.V. Role of interfacial structure on exchange-biased FeF2-Fe. Phys. Rev. B 1999, 59, 6984–6993. [Google Scholar] [CrossRef]
- Renk, O.; Pippan, R. Saturation of grain refinement during severe plastic deformation of single phase materials: Reconsiderations, current status and open questions. Mater. Trans. 2019, 60, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Bachmaier, A.; Schmauch, J.; Aboulfadl, H.; Verch, A.; Motz, C. On the process of co-deformation and phase dissolution in a hard-soft immiscible Cu-Co alloy system during high-pressure torsion deformation. Acta Mater. 2016, 115, 333–346. [Google Scholar] [CrossRef] [Green Version]
- Sabirov, I.; Pippan, R. Characterization of tungsten fragmentation in a W–25%Cu composite after high-pressure torsion. Mater. Charact. 2007, 58, 848–853. [Google Scholar] [CrossRef]
- Groves, G.W.; Kelly, A. Independent slip systems in crystals. Philos. Mag. 1963, 8, 877–887. [Google Scholar] [CrossRef]
- Guiberteau, F.; Donminguez-Rodriguez, A.; Spendel, M.; Castaing, J. Plastic deformation of bunsenite (NiO) at temperatures below 1 050 °C. Rev. Phys. Appliquée 1986, 21, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.G.; Rajdev, D.; Douglass, D.L. The mechanical properties of nickel oxide and their relationship to the morphology of thick oxide scales formed on nickel. Oxid. Met. 1972, 4, 151–170. [Google Scholar] [CrossRef]
- Guétaz, L.; Pénisson, J. High resolution electron microscopy of a deformed superalloy: Shear of precipitates. Mater. Sci. Eng. A 1994, 175, 141–148. [Google Scholar] [CrossRef]
- Sinclair, C.; Embury, J.; Weatherly, G. Basic aspects of the co-deformation of bcc/fcc materials. Mater. Sci. Eng. A 1999, 272, 90–98. [Google Scholar] [CrossRef]
- Wenk, H.R.; Matthies, S.; Hemley, R.J.; Mao, H.K.; Shu, J. The plastic deformation of iron at pressures of the Earth’s inner core. Nature 2000, 405, 1044–1047. [Google Scholar] [CrossRef]
- Kang, F.; Liu, J.Q.; Wang, J.T.; Zhao, X. The effect of hydrostatic pressure on the activation of non-basal slip in a magnesium alloy. Scr. Mater. 2009, 61, 844–847. [Google Scholar] [CrossRef]
- Raabe, D.; Hangen, U. Observation of amorphous areas in a heavily cold rolled Cu-20 wt% Nb composite. Mater. Lett. 1995, 22, 155–161. [Google Scholar] [CrossRef]
- Cabanas-Polo, S.; Bermejo, R.; Ferrari, B.; Sanchez-Herencia, A.J. Ni-NiO composites obtained by controlled oxidation of green compacts. Corros. Sci. 2012, 55, 172–179. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences, and Uses; Wiley-VCH: Hoboken, NJ, USA, 2003; p. 187, 382, 664. [Google Scholar]
- Gaskell, D.R. Discussion of “Representation of mixed reactive gases on free energy (Ellingham-Richardson) diagrams”. Metall. Mater. Trans. B 1996, 27, 693. [Google Scholar] [CrossRef]
- Wriedt, H. The Fe-O (Iron-Oxygen) System. J. Phase Equilibria 1991, 12, 170–200. [Google Scholar] [CrossRef]
- Caruntu, D.; Caruntu, G.; O’Connor, C.J. Magnetic properties of variable-sized Fe3O4 nanoparticles synthesized from non-aqueous homogeneous solutions of polyols. J. Phys. D Appl. Phys. 2007, 40, 5801–5809. [Google Scholar] [CrossRef] [Green Version]
- Roca, A.G.; Marco, J.F.; Del Puerto Morales, M.; Serna, C.J. Effect of nature and particle size on properties of uniform magnetite and maghemite nanoparticles. J. Phys. Chem. C 2007, 111, 18577–18584. [Google Scholar] [CrossRef]
- Berkowitz, A.; Takano, K. Exchange anisotropy—A review. J. Magn. Magn. Mater. 1999, 200, 552–570. [Google Scholar] [CrossRef]
- Berkowitz, A.E.; Schuele, W.J.; Flanders, P.J. Influence of crystallite size on the magnetic properties of acicular γ-Fe2O3 particles. J. Appl. Phys. 1968, 39, 1261–1263. [Google Scholar] [CrossRef]
- Crangle, J.; Goodman, G.M. The magnetization of pure iron and nickel. Proc. R. Soc. Lond. A Math. Phys. Sci. 1971, 321, 477–491. [Google Scholar] [CrossRef]
- Machala, L.; Zboril, R.; Gedanken, A. Amorphous iron(III) oxide—A review. J. Phys. Chem. B 2007, 111, 4003–4018. [Google Scholar] [CrossRef]
- Sauvage, X.; Renaud, L.; Deconihout, B.; Blavette, D.; Ping, D.; Hono, K. Solid state amorphization in cold drawn Cu/Nb wires. Acta Mater. 2001, 49, 389–394. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| <DFe> /[nm] | DFe-WH/[nm] | Fe-WH | <DNiO > /[nm] | DNiO-WH/[nm] | NiO-WH | |
|---|---|---|---|---|---|---|
| Fe50NiO50 @Tdef = RT | 11 ± 2 | 40 | 2 × 10 | 11 ± 2 | 30 | 2 × 10 |
| <DFeNi> / [nm] | DFeNi-WH/[nm] | FeNi-WH | <DFe3O4> /[nm] | DFe3O4-WH/[nm] | Fe3O4-WH | |
| Fe50NiO50 @Tdef = 300 °C | 12 ± 2 | 45 | 3 × 10 | 19 ± 7 | 43 | 1 × 10 |
| <DNi> /[nm] | DNi-WH/[nm] | Ni-WH | <DNiO> /[nm] | DNiO-WH/[nm] | NiO-WH | <D-Fe2O3> /[nm] | |
|---|---|---|---|---|---|---|---|
| vM∼270 | 18 ± 2 | 21 | 3 × 10 | 11 ± 3 | 25 | 2 × 10 | 12 ± 2 |
| vM∼1280 | 15 ± 5 | 19 | 2 × 10 | 11 ± 2 | 20 | 1 × 10 | 9 ± 4 |
| vM∼2560 | 15 ± 4 | 18 | 2 × 10 | 11 ± 2 | 19 | 1 × 10 | 6 ± 2 |
| ann. 450 °C | 21 ± 2 | 22 | 1 × 10 | 14 ± 3 | 22 | 9 × 10 | 8 ± 2 |
| ann. 550 °C | 24 ± 2 | 25 | 0.5 × 10 | 17 ± 3 | 26 | 7 × 10 | 14 ± 2 |
| Heb/[Oe] | HC/[Oe] | HC1/[Oe] | HC2/[Oe] | MS/[emu/g] | MR/[emu/g] | |
|---|---|---|---|---|---|---|
| vM∼90 | −52 | 460 | −512 | 408 | 37.3 | 18.6 |
| vM∼270 | −90 | 625 | −715 | 535 | 32.6 | 21.2 |
| vM∼610 | −124 | 587 | −710 | 463 | 34.1 | 23.3 |
| vM∼1280 | −186 | 786 | −971 | 600 | 28.8 | 23.7 |
| vM∼2560 | −243 | 843 | −1085 | 599 | 27.6 | 23.7 |
| vM∼ 1150 | −200 | 697 | −894 | 494 | 32.3 | 26.8 |
| ann. 450 °C | −67 | 515 | −581 | 449 | 35.8 | 27.0 |
| ann. 550 °C | −23 | 373 | −396 | 349 | 36.6 | 19.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zawodzki, M.; Weissitsch, L.; Krenn, H.; Wurster, S.; Bachmaier, A. Exchange Bias Demonstrated in Bulk Nanocomposites Processed by High-Pressure Torsion. Nanomaterials 2023, 13, 344. https://doi.org/10.3390/nano13020344
Zawodzki M, Weissitsch L, Krenn H, Wurster S, Bachmaier A. Exchange Bias Demonstrated in Bulk Nanocomposites Processed by High-Pressure Torsion. Nanomaterials. 2023; 13(2):344. https://doi.org/10.3390/nano13020344
Chicago/Turabian StyleZawodzki, Michael, Lukas Weissitsch, Heinz Krenn, Stefan Wurster, and Andrea Bachmaier. 2023. "Exchange Bias Demonstrated in Bulk Nanocomposites Processed by High-Pressure Torsion" Nanomaterials 13, no. 2: 344. https://doi.org/10.3390/nano13020344