
Forced Mineral Carbonation of MgO Nanoparticles Synthesized by Aerosol Methods at Room Temperature


Abstract
:1. Introduction
2. Materials and Methods
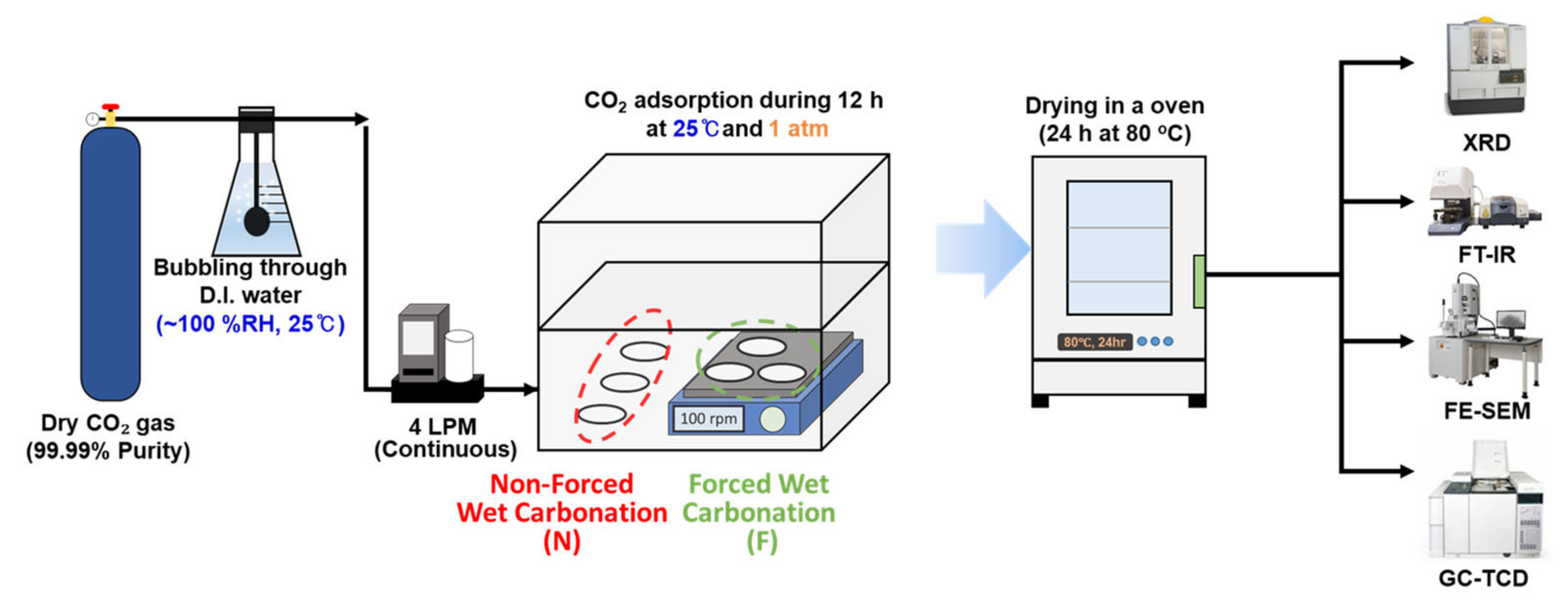
2.1. Preparation and Wet Carbonation Experiments of MgO Nanoparticles
2.2. Analyses of MgO Nanoparticles and the Carbonate Products
2.3. Calculation of Adsorbed CO2 Mass per Unit Mass of Carbonation Samples
3. Results and Discussion
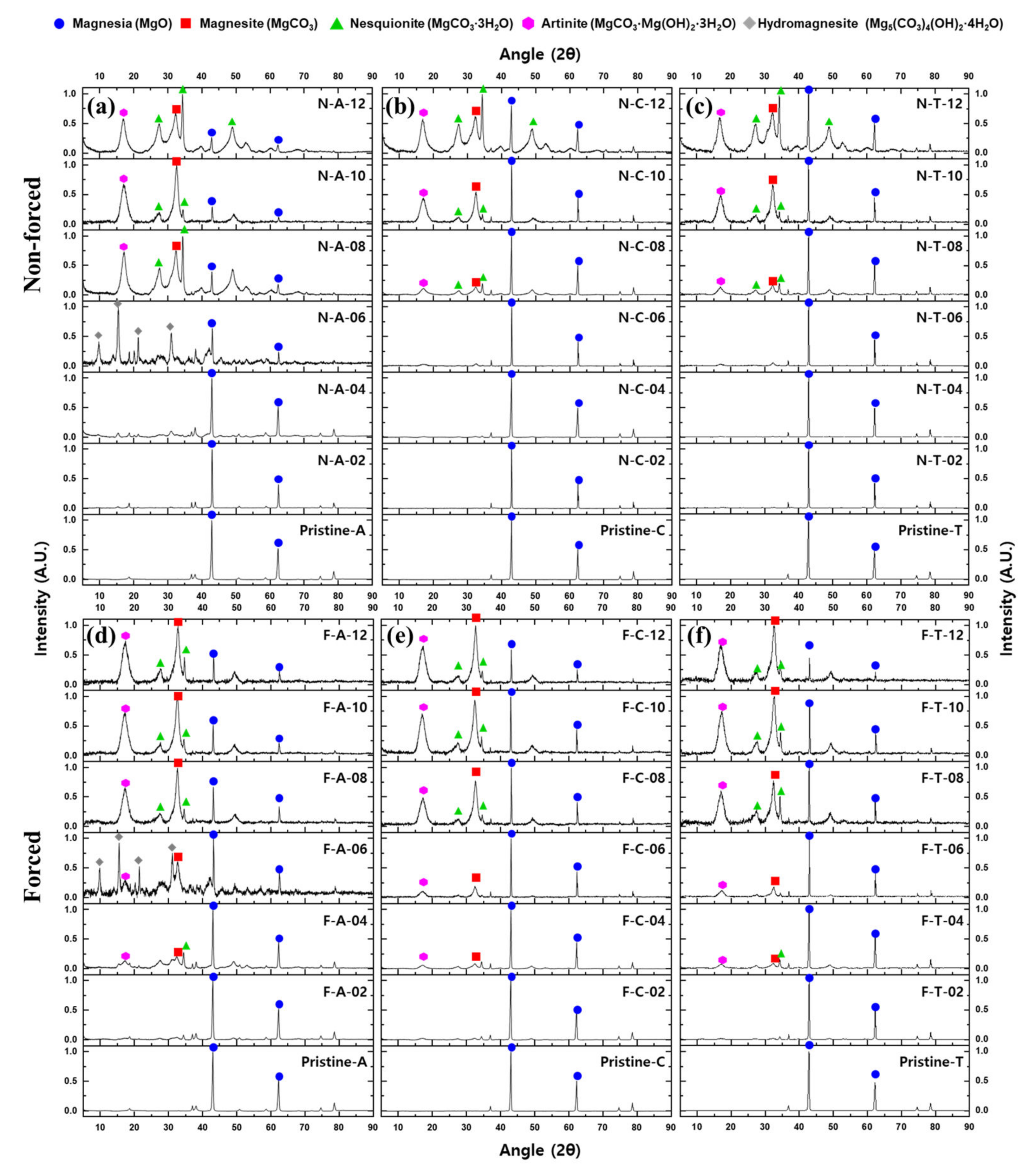
3.1. XRD Result and Crystallinity of Carbonated Samples
3.2. Morphological Changes in MgO Nano-Adsorbents
3.3. FT-IR Result with Different Functional Groups
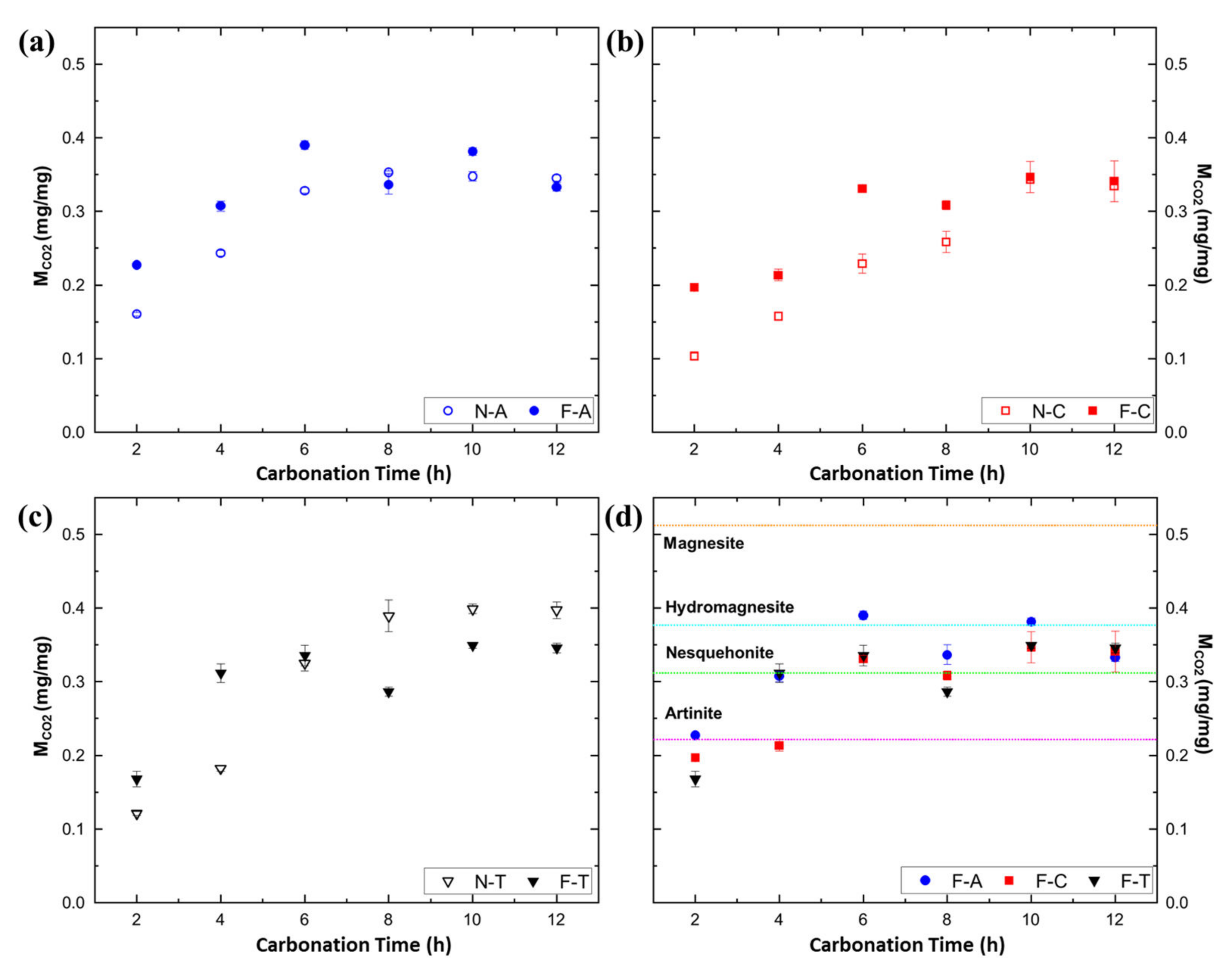
3.4. Quantitative Comparison of MCO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lesk, C.; Rowhani, P.; Ramankutty, N. Influence of Extreme Weather Disasters on Global Crop Production. Nature 2016, 529, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleussner, C.F.; Rogelj, J.; Schaeffer, M.; Lissner, T.; Licker, R.; Fischer, E.M.; Knutti, R.; Levermann, A.; Frieler, K.; Hare, W. Science and Policy Characteristics of the Paris Agreement Temperature Goal. Nat. Clim. Chang. 2016, 6, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Velazquez Abad, A.; Dodds, P.E. Green Hydrogen Characterisation Initiatives: Definitions, Standards, Guarantees of Origin, and Challenges. Energy Policy 2020, 138, 111300. [Google Scholar] [CrossRef]
- Terlouw, T.; Bauer, C.; McKenna, R.; Mazzotti, M. Large-Scale Hydrogen Production via Water Electrolysis: A Techno-Economic and Environmental Assessment. Energy Environ. Sci. 2022, 15, 3583–3602. [Google Scholar] [CrossRef]
- Ajanovic, A.; Sayer, M.; Haas, R. The Economics and the Environmental Benignity of Different Colors of Hydrogen. Int. J. Hydrog. Energy 2022, 47, 24136–24154. [Google Scholar] [CrossRef]
- Cichosz, M.; Kiełkowska, U.; Skowron, K.; Kiedzik, Ł.; Łazarski, S.; Szkudlarek, M.; Kowalska, B.; Żurawski, D. Changes in Synthetic Soda Ash Production and Its Consequences for the Environment. Materials 2022, 15, 4828. [Google Scholar] [CrossRef]
- Pedraza, J.; Zimmermann, A.; Tobon, J.; Schomäcker, R.; Rojas, N. On the Road to Net Zero-Emission Cement: Integrated Assessment of Mineral Carbonation of Cement Kiln Dust. Chem. Eng. J. 2021, 408, 127346. [Google Scholar] [CrossRef]
- Cho, K.; Kim, C. Enhanced Mineral Carbonation at Room Temperature through MgO Nanocubes Synthesized by Self-Combustion. J. Environ. Chem. Eng. 2021, 9, 105592. [Google Scholar] [CrossRef]
- Du, B.; Tandoc, M.C.; Mack, M.L.; Siegel, J.A. Indoor CO2 Concentrations and Cognitive Function: A Critical Review. Indoor Air 2020, 30, 1067–1082. [Google Scholar] [CrossRef]
- Harada, T.; Simeon, F.; Hamad, E.Z.; Hatton, T.A. Alkali Metal Nitrate-Promoted High-Capacity MgO Adsorbents for Regenerable CO2 Capture at Moderate Temperatures. Chem. Mater. 2015, 27, 1943–1949. [Google Scholar] [CrossRef]
- Mosqueda, H.A.; Vazquez, C.; Bosch, P.; Pfeiffer, H. Chemical Sorption of Carbon Dioxide (CO2) on Lithium Oxide (Li2O). Chem. Mater. 2006, 18, 2307–2310. [Google Scholar] [CrossRef]
- Donat, F.; Müller, C.R. Prospects of MgO-Based Sorbents for CO2 Capture Applications at High Temperatures. Curr. Opin. Green Sustain. Chem. 2022, 36, 100645. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, J.; Zhang, Y.; Gao, W.; Harada, T.; Huang, L.; Hatton, T.A.; Wang, Q. Alkali Nitrates Molten Salt Modified Commercial MgO for Intermediate-Temperature CO2 Capture: Optimization of the Li/Na/K Ratio. Ind. Eng. Chem. Res. 2017, 56, 1509–1517. [Google Scholar] [CrossRef]
- Ruminski, A.M.; Jeon, K.J.; Urban, J.J. Size-Dependent CO2 Capture in Chemically Synthesized Magnesium Oxide Nanocrystals. J. Mater. Chem. 2011, 21, 11486–11491. [Google Scholar] [CrossRef]
- Elvira, G.B.; Francisco, G.C.; Víctor, S.M.; Alberto, M.L.R. MgO-Based Adsorbents for CO2 Adsorption: Influence of Structural and Textural Properties on the CO2 Adsorption Performance. J. Environ. Sci. 2017, 57, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Brown, P.; Hatton, T.A. Nonvolatile Colloidal Dispersion of MgO Nanoparticles in Molten Salts for Continuous CO2 Capture at Intermediate Temperatures. ACS Sustain. Chem. Eng. 2019, 7, 7979–7986. [Google Scholar] [CrossRef]
- Hiremath, V.; Hwang, S.; Seo, J.G. Enhanced Cyclic Stability and CO2 Capture Performance of MgO-Al2O3 Sorbent Decorated with Eutectic Mixture. Energy Procedia 2017, 114, 2421–2428. [Google Scholar] [CrossRef]
- Pang, H.; Sun, A.; Xu, H.; Xiao, G. Regenerable MgO-Based Sorbents for CO2 Capture at Elevated Temperature and Pressure: Experimental and DFT Study. Chem. Eng. J. 2021, 425, 130675. [Google Scholar] [CrossRef]
- Song, G.; Zhu, X.; Chen, R.; Liao, Q.; Ding, Y.D.; Chen, L. An Investigation of CO2 Adsorption Kinetics on Porous Magnesium Oxide. Chem. Eng. J. 2016, 283, 175–183. [Google Scholar] [CrossRef]
- Han, S.J.; Bang, Y.; Kwon, H.J.; Lee, H.C.; Hiremath, V.; Song, I.K.; Seo, J.G. Elevated Temperature CO2 Capture on Nano-Structured MgO-Al2O3 Aerogel: Effect of Mg/Al Molar Ratio. Chem. Eng. J. 2014, 242, 357–363. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, T.; Wang, Q. Controlled Synthesis of MgO with Diverse Basic Sites and Its CO2 Capture Mechanism under Different Adsorption Conditions. Chem. Eng. J. 2018, 336, 710–720. [Google Scholar] [CrossRef]
- Lv, G.; Zhu, C.; Zhang, H.; Su, Y.; Qian, P. Mechanism of CO2 Adsorption on Point-Defective MgO Surfaces: First-Principles Study. Appl. Surf. Sci. 2022, 604, 154647. [Google Scholar] [CrossRef]
- Yang, S.; Jang, Y.H.; Kim, C.H.; Hwang, C.; Lee, J.; Chae, S.; Jung, S.; Choi, M. A Flame Metal Combustion Method for Production of Nanoparticles. Powder Technol. 2010, 197, 170–176. [Google Scholar] [CrossRef]
- Pikhitsa, P.V.; Kim, C.; Chae, S.; Shin, S.; Jung, S.; Kitaura, M.; Kimura, S.I.; Fukui, K.; Choi, M. Two-Band Luminescence from an Intrinsic Defect in Spherical and Terraced MgO Nanoparticles. Appl. Phys. Lett. 2015, 106, 183106. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Lee, H.; Pikhitsa, P.V.; Kim, C.; Shin, S.; Kim, D.H.; Choi, M. Synthesis of Terraced and Spherical MgO Nanoparticles Using Flame Metal Combustion. Powder Technol. 2017, 305, 132–140. [Google Scholar] [CrossRef]
- Linga, P.; Kumar, R.; Lee, J.D.; Ripmeester, J.; Englezos, P. A New Apparatus to Enhance the Rate of Gas Hydrate Formation: Application to Capture of Carbon Dioxide. Int. J. Greenh. Gas Control 2010, 4, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Pikhitsa, P.V.; Chae, S.; Cho, K.; Choi, M. Light Emission Induced by Electric Current at Room Temperature through the Defect Networks of MgO Nanocubes. AIP Adv. 2019, 9, 125305. [Google Scholar] [CrossRef]
- Farajzadeh, R.; Zitha, P.L.J.; Bruining, H. Enhanced Mass Transfer of CO2 into Water: Experiment and Modeling. Ind. Eng. Chem. Res. 2009, 48, 6423–6431. [Google Scholar] [CrossRef]
- Van Veelen, A.; Copping, R.; Law, G.T.W.; Smith, A.J.; Bargar, J.R.; Rogers, J.; Shuh, D.K.; Wogelius, R.A. Uranium Uptake onto Magnox Sludge Minerals Studied Using EXAFS. Mineral. Mag. 2012, 76, 3095–3104. [Google Scholar] [CrossRef] [Green Version]
- Ferrini, V.; De Vito, C.; Mignardi, S. Synthesis of Nesquehonite by Reaction of Gaseous CO2 with Mg Chloride Solution: Its Potential Role in the Sequestration of Carbon Dioxide. J. Hazard. Mater. 2009, 168, 832–837. [Google Scholar] [CrossRef]
- Prigiobbe, V.; Mazzotti, M. Precipitation of Mg-Carbonates at Elevated Temperature and Partial Pressure of CO2. Chem. Eng. J. 2013, 223, 755–763. [Google Scholar] [CrossRef]
- Dung, N.T.; Unluer, C. Development of MgO Concrete with Enhanced Hydration and Carbonation Mechanisms. Cem. Concr. Res. 2018, 103, 160–169. [Google Scholar] [CrossRef]
- Hänchen, M.; Prigiobbe, V.; Baciocchi, R.; Mazzotti, M. Precipitation in the Mg-Carbonate System-Effects of Temperature and CO2 Pressure. Chem. Eng. Sci. 2008, 63, 1012–1028. [Google Scholar] [CrossRef]
- Yoo, Y.; Kang, D.; Park, J. Advanced Pseudopolymorph Control of Magnesium Carbonates Using Structural Properties of Amines for CO2 Utilization Based on Post-Treatment of Desalinated Brine. Desalination 2021, 505, 114904. [Google Scholar] [CrossRef]
- Kuenzel, C.; Zhang, F.; Ferrándiz-Mas, V.; Cheeseman, C.R.; Gartner, E.M. The Mechanism of Hydration of MgO-Hydromagnesite Blends. Cem. Concr. Res. 2018, 103, 123–129. [Google Scholar] [CrossRef]
- Choudhari, B.P.; Vaidya, M.C.; Datar, D.S. Physico-chemical studies on basic magnesium carbonates. Indian J. Chem. 1972, 10, 731–733. [Google Scholar]
- Han, H.; Hu, S.; Feng, J.; Gao, H. Effect of Stearic Acid, Zinc Stearate Coating on the Properties of Synthetic Hydromagnesite. Appl. Surf. Sci. 2011, 257, 2677–2682. [Google Scholar] [CrossRef]
- Raade, G. Dypingite, a New Hydrous Basic Carbonate of Magnesium, from Norway. Am. Mineral. 1970, 55, 1457–1465. [Google Scholar]
- White, B.W. Infrared Characterization of Water and Hydroxyl Ion in the Basic Magnesium Carbonate Minerals. Am. Mineral. 1971, 56, 46–53. [Google Scholar]
- Kim, T.K.; Lee, K.J.; Yuh, J.; Kwak, S.K.; Moon, H.R. Multi-Core MgO NPs@C Core-Shell Nanospheres for Selective CO2 Capture under Mild Conditions. New J. Chem. 2014, 38, 1606–1610. [Google Scholar] [CrossRef]
- Kim, T.K.; Lee, K.J.; Cheon, J.Y.; Lee, J.H.; Joo, S.H.; Moon, H.R. Nanoporous Metal Oxides with Tunable and Nanocrystalline Frameworks via Conversion of Metal-Organic Frameworks. J. Am. Chem. Soc. 2013, 135, 8940–8946. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A-MgO (Commercial Nanopowder) | C-MgO (Self-Combustion) | T-MgO (Flame Metal Combustion) | |
|---|---|---|---|
| Surface Area (m2/g−1) | 13.13 | 10.94 | 4.059 |
| Total Pore Volume (cm3/g−1) | 0.1119 | 0.02878 | 0.02716 |
| Average Pore Diameter (nm) | 34.10 | 10.52 | 26.76 |
| (Unit: nm) | A-MgO (Commercial Nanopowder) | C-MgO (Self-Combustion) | T-MgO (Flame Metal Combustion) | |||
|---|---|---|---|---|---|---|
| Carbonation Hour (h) | N | F | N | F | N | F |
| 0 (Pristine) | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0 | 17.6 | 0 | 18.6 | 0 | 17.6 |
| 6 | 0 | 10.0 | 0 | - | 0 | 17.6 |
| 8 | 21.1 | 14.5 | 20.0 | 13.9 | 18.6 | 18.6 |
| 10 | 7.2 | 12.7 | 21.3 | 20.0 | 15.1 | 16.7 |
| 12 | 16.7 | 21.3 | 16.8 | 17.8 | 17.6 | 16.7 |
| (Unit: mg/mg) | A-MgO (Commercial Nanopowder) | C-MgO (Self-Combustion) | T-MgO (Flame Metal Combustion) | |||
|---|---|---|---|---|---|---|
| Carbonation Hour (h) | N | F | N | F | N | F |
| 2 | 0.161 ± 0.002 | 0.227 ± 0.002 | 0.102 ± 0.006 | 0.195 ± 0.006 | 0.121 ± 0.003 | 0.168 ± 0.010 |
| ΔMCO2 = 0.067 | ΔMCO2 = 0.093 | ΔMCO2 = 0.047 | ||||
| 4 | 0.243 ± 0.004 | 0.307 ± 0.007 | 0.156 ± 0.004 | 0.212 ± 0.008 | 0.182 ± 0.003 | 0.312 ± 0.013 |
| ΔMCO2 = 0.064 | ΔMCO2 = 0.056 | ΔMCO2 = 0.129 | ||||
| 6 | 0.328 ± 0.004 | 0.390 ± 0.006 | 0.227 ± 0.013 | 0.329 ± 0.005 | 0.325 ± 0.021 | 0.287 ± 0.006 |
| ΔMCO2 = 0.062 | ΔMCO2 = 0.102 | ΔMCO2 = 0.011 | ||||
| 8 | 0.353 ± 0.002 | 0.337 ± 0.013 | 0.257 ± 0.014 | 0.307 ± 0.006 | 0.389 ± 0.021 | 0.287 ± 0.006 |
| ΔMCO2 = −0.016 | ΔMCO2 = 0.050 | ΔMCO2 = −0.103 | ||||
| 10 | 0.348 ± 0.006 | 0.381 ± 0.005 | 0.342 ± 0.004 | 0.345 ± 0.021 | 0.399 ± 0.006 | 0.349 ± 0.004 |
| ΔMCO2 = 0.034 | ΔMCO2 = 0.003 | ΔMCO2 = −0.050 | ||||
| 12 | 0.345 ± 0.004 | 0.332 ± 0.005 | 0.333 ± 0.005 | 0.339 ± 0.027 | 0.397 ± 0.011 | 0.346 ± 0.006 |
| ΔMCO2 = −0.012 | ΔMCO2 = 0.006 | ΔMCO2 = −0.051 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, K.; Kang, Y.; Chae, S.; Kim, C. Forced Mineral Carbonation of MgO Nanoparticles Synthesized by Aerosol Methods at Room Temperature. Nanomaterials 2023, 13, 281. https://doi.org/10.3390/nano13020281
Cho K, Kang Y, Chae S, Kim C. Forced Mineral Carbonation of MgO Nanoparticles Synthesized by Aerosol Methods at Room Temperature. Nanomaterials. 2023; 13(2):281. https://doi.org/10.3390/nano13020281
Chicago/Turabian StyleCho, Kyungil, Yeryeong Kang, Sukbyung Chae, and Changhyuk Kim. 2023. "Forced Mineral Carbonation of MgO Nanoparticles Synthesized by Aerosol Methods at Room Temperature" Nanomaterials 13, no. 2: 281. https://doi.org/10.3390/nano13020281