
Polymer-Based Nanoparticles as Drug Delivery Systems for Purines of Established Importance in Medicine †
 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Polymeric Nanospheres with Purines
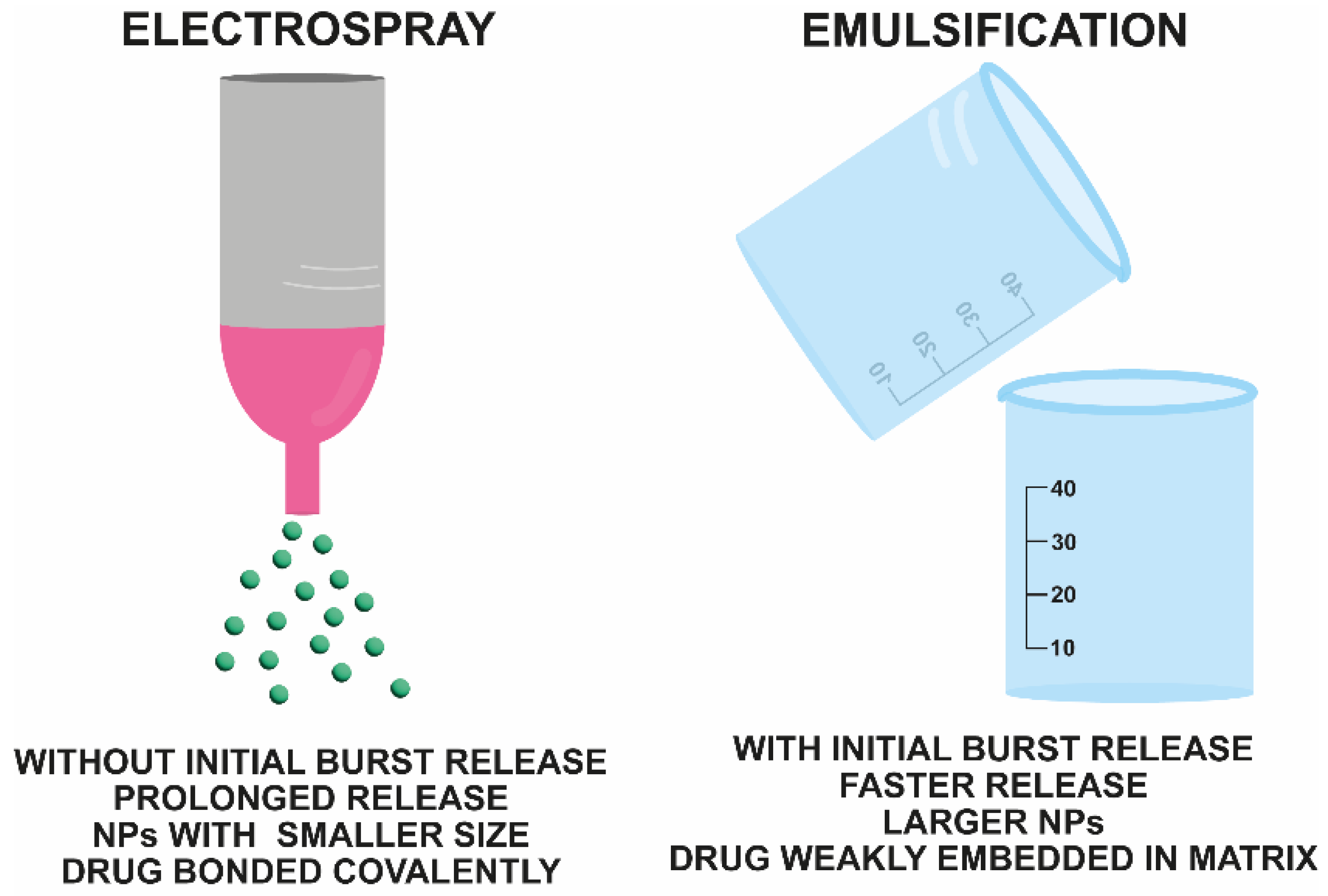
2.1. PLGA Nanospheres
2.1.1. PLGA Nanoparticles with 6-Mercaptopurine and 6-Thioguanine
2.1.2. PLGA Nanoparticles with Acyclovir and Ganciclovir
2.1.3. PLGA Nanoparticles with Tenofovir and Its Prodrugs
2.2. Methacrylate Copolymer-Based Nanospheres
3. Nanogels with Purines
3.1. Chitosan-Based Nanogels

| API | Ø (nm) | ζ (mV) | EE (%) | Summary | Ref. |
|---|---|---|---|---|---|
| 6-MP | 187 | 54.9 | n.g. |
| [92] |
| 90 | 26.2 | n.g. |
| [107] | |
| 137.9 | 53.8 | 29.10 |
| [93] | |
| 6-TG | 261.63 | 15.97 | 44.27 |
| [23] |
| CLA | 636–820 | n.g. | 62 |
| [98] |
| ACV | 220 | 4.1 | 62.5 |
| [6] |
| 200 | 36.7 | 56 |
| [100] | |
| DDI | 382 | n.g. | 94.6 |
| [11] |
| TNF | 315 | n.g. | 49 |
| [106] |
| To sum up | |||||
| Pros | |||||
| |||||
| Cons | |||||
| |||||
3.2. Other Nanogels
3.2.1. Gelatin-Based Nanogels
3.2.2. Poly(ethyleneimine)-Based Nanogels
3.3. Alternative for Nanogels
4. Dendrimers
| API | Dendrimer Material | Generation | Summary | Ref. |
|---|---|---|---|---|
| FLU | poly(propyleneimine) | fourth |
| [129,130] |
| CLO | poly(propyleneimine) | fourth |
| [130] |
| 6-MP | melanin | fourth |
| [137] |
| hydroxyl terminated poly(amidoamine) | fourth |
| [138] | |
| hydroxyl terminated poly(amidoamine) with gold NPs ⌀ 3 nm | fifth |
| [140] | |
| ACV | thiolated poly(amidoamine) | modified third |
| [141] |
| To sum up | ||||
| Pros | ||||
| ||||
| Cons | ||||
| ||||
5. Polymeric Micelles with Purines
| API | Ø (nm) | Summary | Ref. |
|---|---|---|---|
| 6-MP | 160 |
| [142] |
| 55.8 |
| [143] | |
| 116 |
| [144] | |
| 115.8 |
| [147] | |
| 163.3 |
| [145] | |
| 6-TG | 162.4 | ||
| 30 |
| [146] | |
| ACV | 141.8 |
| [149] |
| 172.7 |
| ||
| GCV | 117 |
| [150] |
| To sum up | |||
| Pros | |||
| |||
| Cons | |||
| |||
6. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Brunton, L.L.; Knollmann, B.C.; Hilal-Dandan, R. (Eds.) Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 13th ed.; McGraw Hill Medical: New York, NY, USA, 2018. [Google Scholar]
- Dahiya, S.; Pathak, K.; Sharma, R. Development of Extended Release Coevaporates and Coprecipitates of Promethazine HCl with Acrylic Polymers: Formulation Considerations. Chem. Pharm. Bull. 2008, 56, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Lech-Maranda, E.; Korycka, A.; Robak, T. Pharmacological and Clinical Studies on Purine Nucleoside Analogs- New Anticancer Agents. MRMC 2006, 6, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Asberg, A.; Chou, S.; Danziger-Isakov, L.; Humar, A. Transplantation Society International CMV Consensus Group Updated International Consensus Guidelines on the Management of Cytomegalovirus in Solid-Organ Transplantation. Transplantation 2013, 96, 333–360. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, P.; García-Benayas, T.; Rendón, A.; Rodríguez-Novoa, S.; Soriano, V. Combinations of Nucleoside/Nucleotide Analogues for HIV Therapy. AIDS Rev. 2004, 6, 234–243. [Google Scholar]
- Jain, N.; Rajoriya, V.; Jain, P.K.; Jain, A.K. Lactosaminated-N-Succinyl Chitosan Nanoparticles for Hepatocyte-Targeted Delivery of Acyclovir. J. Nanopart. Res. 2014, 16, 2136. [Google Scholar] [CrossRef]
- Alkholief, M.; Albasit, H.; Alhowyan, A.; Alshehri, S.; Raish, M.; Abul Kalam, M.; Alshamsan, A. Employing a PLGA-TPGS Based Nanoparticle to Improve the Ocular Delivery of Acyclovir. Saudi Pharm. J. 2019, 27, 293–302. [Google Scholar] [CrossRef]
- Jwala, J.; Boddu, S.H.S.; Shah, S.; Sirimulla, S.; Pal, D.; Mitra, A.K. Ocular Sustained Release Nanoparticles Containing Stereoisomeric Dipeptide Prodrugs of Acyclovir. J. Ocul. Pharmacol. Ther. 2011, 27, 163–172. [Google Scholar] [CrossRef]
- Gupta, S.; Agarwal, A.; Gupta, N.K.; Saraogi, G.; Agrawal, H.; Agrawal, G.P. Galactose Decorated PLGA Nanoparticles for Hepatic Delivery of Acyclovir. Drug Dev. Ind. Pharm. 2013, 39, 1866–1873. [Google Scholar] [CrossRef]
- Ensign, L.M.; Tang, B.C.; Wang, Y.-Y.; Tse, T.A.; Hoen, T.; Cone, R.; Hanes, J. Mucus-Penetrating Nanoparticles for Vaginal Drug Delivery Protect Against Herpes Simplex Virus. Sci. Transl. Med. 2012, 4, 138ra79. [Google Scholar] [CrossRef]
- Al-Ghananeem, A.M.; Saeed, H.; Florence, R.; Yokel, R.A.; Malkawi, A.H. Intranasal Drug Delivery of Didanosine-Loaded Chitosan Nanoparticles for Brain Targeting; an Attractive Route against Infections Caused by Aids Viruses. J. Drug Target. 2010, 18, 381–388. [Google Scholar] [CrossRef]
- McGovern, D.P.B.; Travis, S.P.L.; Duley, J.; Shobowale-Bakre, E.M.; Dalton, H.R. Azathioprine Intolerance in Patients with IBD May Be Imidazole-Related and Is Independent of TPMT Activity. Gastroenterology 2002, 122, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Liliemark, J. The Clinical Pharmacokinetics of Cladribine. Clin. Pharmacokinet. 1997, 32, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Namiot, E.D.; Sokolov, A.V.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Nanoparticles in Clinical Trials: Analysis of Clinical Trials, FDA Approvals and Use for COVID-19 Vaccines. Int. J. Mol. Sci. 2023, 24, 787. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.V.; Gonçalves, V.; Da Silva, M.C.; Bañobre-López, M.; Gallo, J. PLGA-Based Composites for Various Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 2034. [Google Scholar] [CrossRef] [PubMed]
- Tawade, P.; Tondapurkar, N.; Jangale, A. Biodegradable and Biocompatible Synthetic Polymers for Applications in Bone and Muscle Tissue Engineering. J. Med. Sci. 2022, 91, e712. [Google Scholar] [CrossRef]
- Singh, I.; Rana, V. Iron Oxide Induced Enhancement of Mucoadhesive Potential of Eudragit RLPO: Formulation, Evaluation and Optimization of Mucoadhesive Drug Delivery System. Expert Opin. Drug Deliv. 2013, 10, 1179–1191. [Google Scholar] [CrossRef]
- Pandey, S.; Jirwankar, P.; Mehta, S.; Pandit, S.; Tripathi, P.; Patil, A. Formulation and Evaluation of Bilayered Gastroretentable Mucoadhesive Patch for Stomach-Specific Drug Delivery. CDD 2013, 10, 374–383. [Google Scholar] [CrossRef]
- Sahoo, J.; Murthy, P.N.; Biswal, S. Manik Formulation of Sustained-Release Dosage Form of Verapamil Hydrochloride by Solid Dispersion Technique Using Eudragit RLPO or Kollidon®SR. AAPS PharmSciTech 2009, 10, 27–33. [Google Scholar] [CrossRef]
- Gandhi, A.; Jana, S.; Sen, K.K. In-Vitro Release of Acyclovir Loaded Eudragit RLPO® Nanoparticles for Sustained Drug Delivery. Int. J. Biol. Macromol. 2014, 67, 478–482. [Google Scholar] [CrossRef]
- Palem, C.R.; Gannu, R.; Doodipala, N.; Yamsani, V.V.; Yamsani, M.R. Transmucosal Delivery of Domperidone from Bilayered Buccal Patches: In Vitro, Ex Vivo and In Vivo Characterization. Arch. Pharm. Res. 2011, 34, 1701–1710. [Google Scholar] [CrossRef]
- Varshosaz, J.; Faghihian, H.; Rastgoo, K. Preparation and Characterization of Metoprolol Controlled-Release Solid Dispersions. Drug Deliv. 2006, 13, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Rajashekaraiah, R.; Kumar, P.R.; Prakash, N.; Rao, G.S.; Devi, V.R.; Metta, M.; Narayanaswamy, H.D.; Swamy, M.N.; Satyanarayan, K.; Rao, S.; et al. Anticancer Efficacy of 6-Thioguanine Loaded Chitosan Nanoparticles with or without Curcumin. Int. J. Biol. Macromol. 2020, 148, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Heras Caballero, A.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wang, Y.; Li, J.; Chen, S. Progress and Prospects in Chitosan Derivatives: Modification Strategies and Medical Applications. J. Mater. Sci. Technol. 2021, 89, 209–224. [Google Scholar] [CrossRef]
- Nur Hanani, Z.A.; Roos, Y.H.; Kerry, J.P. Use and Application of Gelatin as Potential Biodegradable Packaging Materials for Food Products. Int. J. Biol. Macromol. 2014, 71, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Luo, Q.; Chu, Y.; Tao, N.; Deng, S.; Wang, L.; Li, L. Application of Gelatin in Food Packaging: A Review. Polymers 2022, 14, 436. [Google Scholar] [CrossRef]
- Saletnik, Ł.; Wesołowski, R. Fluorescent Spectroscopy of Collagen as a Diagnostic Tool in Medicine. J. Med. Sci. 2022, 91, e584. [Google Scholar] [CrossRef]
- Aisina, R.; Mukhametova, L.; Ivanova, E. Influence Cationic and Anionic PAMAM Dendrimers of Low Generation on Selected Hemostatic Parameters in Vitro. Mater. Sci. Eng. C 2020, 109, 110605. [Google Scholar] [CrossRef]
- Ziemba, B.; Janaszewska, A.; Ciepluch, K.; Krotewicz, M.; Fogel, W.A.; Appelhans, D.; Voit, B.; Bryszewska, M.; Klajnert, B. In Vivo Toxicity of Poly(Propyleneimine) Dendrimers. J. Biomed. Mater. Res. 2011, 99A, 261–268. [Google Scholar] [CrossRef]
- Khansarizadeh, M.; Mokhtarzadeh, A.; Rashedinia, M.; Taghdisi, S.; Lari, P.; Abnous, K.; Ramezani, M. Identification of Possible Cytotoxicity Mechanism of Polyethylenimine by Proteomics Analysis. Hum. Exp. Toxicol. 2016, 35, 377–387. [Google Scholar] [CrossRef]
- Kazemi Oskuee, R.; Dabbaghi, M.; Gholami, L.; Taheri-Bojd, S.; Balali-Mood, M.; Mousavi, S.H.; Malaekeh-Nikouei, B. Investigating the Influence of Polyplex Size on Toxicity Properties of Polyethylenimine Mediated Gene Delivery. Life Sci. 2018, 197, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Rellegadla, S.; Jain, S. Biomedical Applications of PLGA Particles. In Materials for Biomedical Engineering; Elsevier: Amsterdam, The Netherlands, 2019; pp. 87–129. ISBN 978-0-12-816913-1. [Google Scholar]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Pereira, M.C. PLGA Based Drug Carrier and Pharmaceutical Applications: The Most Recent Advances. Pharmaceutics 2020, 12, 903. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lv, X.; Le, Y. Chitosan-Modified PLGA Nanoparticles for Control-Released Drug Delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Fan, L.; Zha, Y.; Guo, L.; Zhang, Q.; Chen, J.; Pang, Z.; Wang, Y.; Jiang, X.; et al. Targeting the Brain with PEG–PLGA Nanoparticles Modified with Phage-Displayed Peptides. Biomaterials 2011, 32, 4943–4950. [Google Scholar] [CrossRef]
- Essa, D.; Choonara, Y.E.; Kondiah, P.P.D.; Pillay, V. Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques. Polymers 2020, 12, 1882. [Google Scholar] [CrossRef]
- DrugBank Online. Available online: https://go.drugbank.com/ (accessed on 29 August 2023).
- Yuan, B.; Zhang, J.; Wang, H.; Xiong, L.; Cai, Q.; Wang, T.; Jacobsen, S.; Pradhan, S.; Wang, Y. 6-Thioguanine Reactivates Epigenetically Silenced Genes in Acute Lymphoblastic Leukemia Cells by Facilitating Proteasome-Mediated Degradation of DNMT1. Cancer Res. 2011, 71, 1904–1911. [Google Scholar] [CrossRef]
- Florin, T.H.J.; Wright, J.D.; Jambhrunkar, S.D.; Henman, M.G.; Popat, A. A Well-Tolerated and Rapidly Acting Thiopurine for IBD? Drug Discov. Today 2019, 24, 37–41. [Google Scholar] [CrossRef]
- Chen, D.; Liu, Y.; Zhang, F.; You, Q.; Ma, W.; Wu, J.; Wu, Z. 6-Thioguanine Inhibits Herpes Simplex Virus 1 Infection of Eyes. Microbiol. Spectr. 2021, 9, e00646-21. [Google Scholar] [CrossRef]
- Brem, R.; Karran, P. Oxidation-Mediated DNA Cross-Linking Contributes to the Toxicity of 6-Thioguanine in Human Cells. Cancer Res. 2012, 72, 4787–4795. [Google Scholar] [CrossRef]
- Toksvang, L.N.; Schmidt, M.S.; Arup, S.; Larsen, R.H.; Frandsen, T.L.; Schmiegelow, K.; Rank, C.U. Hepatotoxicity during 6-Thioguanine Treatment in Inflammatory Bowel Disease and Childhood Acute Lymphoblastic Leukaemia: A Systematic Review. PLoS ONE 2019, 14, e0212157. [Google Scholar] [CrossRef] [PubMed]
- Dean, L. Thioguanine Therapy and TPMT and NUDT15 Genotype. In Medical Genetics Summaries; Pratt, V.M., Scott, S.A., Pirmohamed, M., Esquivel, B., Kattman, B.L., Malheiro, A.J., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Zou, Y.; Mei, D.; Yuan, J.; Han, J.; Xu, J.; Sun, N.; He, H.; Yang, C.; Zhao, L. Preparation, Characterization, Pharmacokinetic, and Therapeutic Potential of Novel 6-Mercaptopurine-Loaded Oral Nanomedicines for Acute Lymphoblastic Leukemia. IJN 2021, 16, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Jaiswal, N.; Hens, A.; Mahata, N.; Chanda, N. Development of 6-Thioguanine Conjugated PLGA Nanoparticles through Thioester Bond Formation: Benefits of Electrospray Mediated Drug Encapsulation and Sustained Release in Cancer Therapeutic Applications. Mater. Sci. Eng. C 2020, 114, 111029. [Google Scholar] [CrossRef] [PubMed]
- James, S.H.; Prichard, M.N. Current and Future Therapies for Herpes Simplex Virus Infections: Mechanism of Action and Drug Resistance. Curr. Opin. Virol. 2014, 8, 54–61. [Google Scholar] [CrossRef]
- Elion, G.B. Mechanism of Action and Selectivity of Acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Gold Standard Drug Database—Elsevier. Available online: https://www.elsevier.com/solutions/drug-database (accessed on 5 August 2023).
- Yen, H.L.; Tsai, S.C. Acute Kidney Injury Due to Acyclovir: A Case Report. J. Neurol. Sci. 2017, 381, 1015. [Google Scholar] [CrossRef]
- Brandariz-Nuñez, D.; Correas-Sanahuja, M.; Maya-Gallego, S.; Martín Herranz, I. Neurotoxicity Associated with Acyclovir and Valacyclovir: A Systematic Review of Cases. J. Clin. Pharm. Ther. 2021, 46, 918–926. [Google Scholar] [CrossRef]
- The University of Cincinnati Residents; Makley, A. The Mont Reid Surgical Handbook, 7th ed.; Elsevier: Philadelphia, PA, USA; ISBN 9780323529808. Available online: https://www.asia.elsevierhealth.com/the-mont-reid-surgical-handbook-9780323529808.html (accessed on 5 August 2023).
- Spector, S.A.; Weingeist, T.; Pollard, R.B.; Dieterich, D.T.; Sarno, T.; Benson, C.A.; Busch, D.F.; Freeman, W.R.; Montague, P.; Kaplan, H.J.; et al. A Randomized, Controlled Study of Intravenous Ganciclovir Therapy for Cytomegalovirus Peripheral Retinitis in Patients with AIDS. J. Infect. Dis. 1993, 168, 557–563. [Google Scholar] [CrossRef]
- Razonable, R.R. Antiviral Drugs for Viruses Other than Human Immunodeficiency Virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef]
- Central Nervous System Side Effects of Ganciclovir. N. Engl. J. Med. 1990, 322, 933–934. [CrossRef]
- Combarnous, F.; Fouque, D.; Chossegros, P.; Boulieu, R.; Laville, M.; Zech, P. Neurologic Side-Effects of Ganciclovir. Clin. Nephrol. 1994, 42, 279–280. [Google Scholar]
- Kamel, A.O.; Awad, G.A.S.; Geneidi, A.S.; Mortada, N.D. Preparation of Intravenous Stealthy Acyclovir Nanoparticles with Increased Mean Residence Time. AAPS PharmSciTech 2009, 10, 1427. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic Modeling on Drug Release from Controlled Drug Delivery Systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Bhosale, U.V.; Kusum Devi, V.; Jain, N. Formulation and Optimization of Mucoadhesive Nanodrug Delivery System of Acyclovir. J. Young Pharm. 2011, 3, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kucers’ the Use of Antibiotics: A Clinical Review of Antibacterial, Antifungal, Antiparasitic and Antiviral Drugs, 7th ed.; Grayson, M.L. (Ed.) CRC Press: Boca Raton, FL, USA, 2017; ISBN 978-1-4987-4795-0. [Google Scholar]
- Badoni, A.; Ojha, A.; Gnanarajan, G.; Kothiyal, P. Review on Gastro Retentive Drug Delivery System. Pharma Innov. 2012, 1, 11. [Google Scholar]
- Al-Dhubiab, B.E.; Nair, A.B.; Kumria, R.; Attimarad, M.; Harsha, S. Formulation and Evaluation of Nano Based Drug Delivery System for the Buccal Delivery of Acyclovir. Colloids Surf. B Biointerfaces 2015, 136, 878–884. [Google Scholar] [CrossRef]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A Review of the Toxicity of HIV Medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef]
- De Clercq, E. The Holý Trinity: The Acyclic Nucleoside Phosphonates. Adv. Pharmacol. 2013, 67, 293–316. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A Review: Mechanism of Action of Antiviral Drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Lou, L. Advances in Nucleotide Antiviral Development from Scientific Discovery to Clinical Applications: Tenofovir Disoproxil Fumarate for Hepatitis B. JCTH 2013, 1, 33. [Google Scholar] [CrossRef]
- Quimby, D.; Brito, M.O. Fanconi Syndrome Associated with Use of Tenofovir in HIV-Infected Patients: A Case Report and Review of the Literature. AIDS Read 2005, 15, 357–364. [Google Scholar] [PubMed]
- Mangioni, D.; Bandera, A.; Muscatello, A.; Squillace, N.; Crivellaro, C.; Guerra, L.; Messa, C.; Gori, A. Focal Bone Lesions in Hiv-Positive Patient Treated with Tenofovir. BMC Infect. Dis. 2014, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Lucey, J.M.; Hsu, P.; Ziegler, J.B. Tenofovir-Related Fanconi’s Syndrome and Osteomalacia in a Teenager with HIV. BMJ Case Rep. 2013, 2013, bcr2013008674. [Google Scholar] [CrossRef]
- Van Rompay, K.K.A.; Brignolo, L.L.; Meyer, D.J.; Jerome, C.; Tarara, R.; Spinner, A.; Hamilton, M.; Hirst, L.L.; Bennett, D.R.; Canfield, D.R.; et al. Biological Effects of Short-Term or Prolonged Administration of 9-[2-(Phosphonomethoxy)Propyl]Adenine (Tenofovir) to Newborn and Infant Rhesus Macaques. Antimicrob. Agents Chemother. 2004, 48, 1469–1487. [Google Scholar] [CrossRef]
- Zhang, T.; Sturgis, T.F.; Youan, B.-B.C. pH-Responsive Nanoparticles Releasing Tenofovir Intended for the Prevention of HIV Transmission. Eur. J. Pharm. Biopharm. 2011, 79, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Reis, C.; Machado, A.; Barreiros, L.; Araújo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; Segundo, M.A.; Sarmento, B.; das Neves, J. Nanoparticles-in-Film for the Combined Vaginal Delivery of Anti-HIV Microbicide Drugs. J. Control. Release 2016, 243, 43–53. [Google Scholar] [CrossRef]
- Machado, A.; Cunha-Reis, C.; Araújo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; das Neves, J.; Sarmento, B. Development and in Vivo Safety Assessment of Tenofovir-Loaded Nanoparticles-in-Film as a Novel Vaginal Microbicide Delivery System. Acta Biomater. 2016, 44, 332–340. [Google Scholar] [CrossRef]
- Destache, C.J.; Mandal, S.; Yuan, Z.; Kang, G.; Date, A.A.; Lu, W.; Shibata, A.; Pham, R.; Bruck, P.; Rezich, M.; et al. Topical Tenofovir Disoproxil Fumarate Nanoparticles Prevent HIV-1 Vaginal Transmission in a Humanized Mouse Model. Antimicrob. Agents Chemother. 2016, 60, 3633–3639. [Google Scholar] [CrossRef]
- Belletti, D.; Tosi, G.; Forni, F.; Gamberini, M.C.; Baraldi, C.; Vandelli, M.A.; Ruozi, B. Chemico-Physical Investigation of Tenofovir Loaded Polymeric Nanoparticles. Int. J. Pharm. 2012, 436, 753–763. [Google Scholar] [CrossRef]
- Shailender, J.; Ravi, P.R.; Saha, P.; Dalvi, A.; Myneni, S. Tenofovir Disoproxil Fumarate Loaded PLGA Nanoparticles for Enhanced Oral Absorption: Effect of Experimental Variables and in Vitro, Ex Vivo and in Vivo Evaluation. Colloids Surf. B Biointerfaces 2017, 158, 610–619. [Google Scholar] [CrossRef]
- Patra, C.N.; Priya, R.; Swain, S.; Kumar Jena, G.; Panigrahi, K.C.; Ghose, D. Pharmaceutical Significance of Eudragit: A Review. Future J. Pharm. Sci. 2017, 3, 33–45. [Google Scholar] [CrossRef]
- Porwal, A.; Swami, G.; Saraf, S. Preparation and Evaluation of Sustained Release Microballoons of Propranolol. Daru 2011, 19, 193–201. [Google Scholar] [PubMed]
- Jalil, R.; Roni, M.; Kibria, G. Formulation and in Vitro Evaluation of Alfuzosin Extended Release Tablets Using Directly Compressible Eudragit. Indian J. Pharm. Sci. 2009, 71, 252. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Dhal, S.; Mohapatro, P.; Behera, B.C.H.; Barik, B.B. Effect of Processing Temperature on Eudragit RS PO Microsphere Characteristics in the Solvent Evaporation Process. Pharmazie 2007, 62, 638–639. [Google Scholar] [CrossRef]
- Boza, A.; Caraballo, I.; Alvarez-Fuentes, J.; Rabasco, A.M. Evaluation of Eudragit® RS-PO and Ethocel® 100 Matrices for the Controlled Release of Lobenzarit Disodium. Drug Dev. Ind. Pharm. 1999, 25, 229–233. [Google Scholar] [CrossRef]
- Rey, H.; Wagner, K.; Wehrlé, P.; Schmidt, P. Development of Matrix-Based Theophylline Sustained-Release Microtablets. Drug Dev. Ind. Pharm. 2000, 26, 21–26. [Google Scholar] [CrossRef]
- Ramyadevi, D.; Rajan, K.S.; Vedhahari, B.N.; Ruckmani, K.; Subramanian, N. Heterogeneous Polymer Composite Nanoparticles Loaded in Situ Gel for Controlled Release Intra-Vaginal Therapy of Genital Herpes. Colloids Surf. B Biointerfaces 2016, 146, 260–270. [Google Scholar] [CrossRef]
- Aminabhavi, T.M.; Dharupaneedi, S.P.; More, U.A. The Role of Nanotechnology and Chitosan-Based Biomaterials for Tissue Engineering and Therapeutic Delivery. In Chitosan Based Biomaterials Volume 2; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–29. ISBN 978-0-08-100228-5. [Google Scholar]
- Pérez-Álvarez, L.; Ruiz-Rubio, L.; Vilas-Vilela, J.L. Determining the Deacetylation Degree of Chitosan: Opportunities To Learn Instrumental Techniques. J. Chem. Educ. 2018, 95, 1022–1028. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, C.; Wang, X.; Fang, Q.; Huang, J. Synthesis, Characterization, and Antimicrobial Activities of Sulfonated Chitosan. Carbohydr. Polym. 2017, 155, 321–328. [Google Scholar] [CrossRef]
- Costa, E.M.; Silva, S.; Pina, C.; Tavaria, F.K.; Pintado, M.M. Evaluation and Insights into Chitosan Antimicrobial Activity against Anaerobic Oral Pathogens. Anaerobe 2012, 18, 305–309. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar] [CrossRef]
- Nguyen, S.; Hisiger, S.; Jolicoeur, M.; Winnik, F.M.; Buschmann, M.D. Fractionation and Characterization of Chitosan by Analytical SEC and 1H NMR after Semi-Preparative SEC. Carbohydr. Polym. 2009, 75, 636–645. [Google Scholar] [CrossRef]
- Arata Badano, J.; Vanden Braber, N.; Rossi, Y.; Díaz Vergara, L.; Bohl, L.; Porporatto, C.; Falcone, R.D.; Montenegro, M. Physicochemical, in Vitro Antioxidant and Cytotoxic Properties of Water-Soluble Chitosan-Lactose Derivatives. Carbohydr. Polym. 2019, 224, 115158. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Joladarashi, D.; Hallur, R.L.S.; Sanganal, J.S.; Phani, A.R. Toxicity Evaluation of 6-Mercaptopurine-Chitosan Nanoparticles in Rats. Saudi Pharm. J. 2020, 28, 147–154. [Google Scholar] [CrossRef]
- Kumar, G.P.; Sanganal, J.S.; Phani, A.R.; Manohara, C.; Tripathi, S.M.; Raghavendra, H.L.; Janardhana, P.B.; Amaresha, S.; Swamy, K.B.; Prasad, R.G.S.V. Anti-Cancerous Efficacy and Pharmacokinetics of 6-Mercaptopurine Loaded Chitosan Nanoparticles. Pharmacol. Res. 2015, 100, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Hermann, R.; Krajcsi, P.; Fluck, M.; Seithel-Keuth, A.; Bytyqi, A.; Galazka, A.; Munafo, A. Cladribine as a Potential Object of Nucleoside Transporter-Based Drug Interactions. Clin. Pharmacokinet. 2022, 61, 167–187. [Google Scholar] [CrossRef]
- Kobayashi, K.; Vogelzang, N.J.; O’Brien, S.M.; Schilsky, R.L.; Vokes, E.E.; Ratain, M.J. A Phase I Study of Intermittent Infusion Cladribine in Patients with Solid Tumors. Cancer 1994, 74, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Bloński, J.Z.; Kasznicki, M.; Blasińska-Morawiec, M.; Krykowski, E.; Dmoszyńska, A.; Mrugala-Spiewak, H.; Skotnicki, A.B.; Nowak, W.; Konopka, L.; et al. Cladribine with Prednisone versus Chlorambucil with Prednisone as First-Line Therapy in Chronic Lymphocytic Leukemia: Report of a Prospective, Randomized, Multicenter Trial. Blood 2000, 96, 2723–2729. [Google Scholar] [PubMed]
- Lech-Maranda, E.; Seweryn, M.; Giebel, S.; Holowiecki, J.; Piatkowska-Jakubas, B.; Wegrzyn, J.; Skotnicki, A.; Kielbinski, M.; Kuliczkowski, K.; Paluszewska, M.; et al. Infectious Complications in Patients with Acute Myeloid Leukemia Treated According to the Protocol with Daunorubicin and Cytarabine with or without Addition of Cladribine. A Multicenter Study by the Polish Adult Leukemia Group (PALG). Int. J. Infect. Dis. 2010, 14, e132–e140. [Google Scholar] [CrossRef] [PubMed]
- Domaratzki, R.E.; Ghanem, A. Encapsulation and Release of Cladribine from Chitosan Nanoparticles. J. Appl. Polym. Sci. 2012, 128, 2173–2179. [Google Scholar] [CrossRef]
- Nasrabadi, M.; Morsali, A.; Beyramabadi, S.A. An Applied Quantum-Chemical Model for Genipin-Crosslinked Chitosan (GCS) Nanocarrier. Int. J. Biol. Macromol. 2020, 165, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, N.; Natrajan, R.; Kumar, R.; Selvaraj, S. Acyclovir-Loaded Chitosan Nanoparticles for Ocular Delivery. Asian J. Pharm. 2010, 4, 220. [Google Scholar] [CrossRef]
- Harmening, N.; Klug, M.; Gramann, K.; Miklody, D. HArtMuT—Modeling Eye and Muscle Contributors in Neuroelectric Imaging. J. Neural Eng. 2022, 19, 066041. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Noble, S. Didanosine: An Updated Review of Its Use in HIV Infection. Drugs 1999, 58, 1099–1135. [Google Scholar] [CrossRef] [PubMed]
- Haug, S.J.; Wong, R.W.; Day, S.; Choudhry, N.; Sneed, S.; Prasad, P.; Read, S.; McDonald, R.H.; Agarwal, A.; Davis, J.; et al. DIDANOSINE RETINAL TOXICITY. Retina 2016, 36, S159–S167. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Peripheral Neuropathy and Antiretroviral Drugs. J. Peripher. Nerv. Syst. 2001, 6, 14–20. [Google Scholar] [CrossRef]
- Martin, J.C.; Hitchcock, M.J.M.; De Clercq, E.; Prusoff, W.H. Early Nucleoside Reverse Transcriptase Inhibitors for the Treatment of HIV: A Brief History of Stavudine (D4T) and Its Comparison with Other Dideoxynucleosides. Antiviral. Res. 2010, 85, 34–38. [Google Scholar] [CrossRef]
- Narayanan, V.H.B.; Lewandowski, A.; Durai, R.; Gonciarz, W.; Wawrzyniak, P.; Brzezinski, M. Spray-Dried Tenofovir Alafenamide-Chitosan Nanoparticles Loaded Oleogels as a Long-Acting Injectable Depot System of Anti-HIV Drug. Int. J. Biol. Macromol. 2022, 222, 473–486. [Google Scholar] [CrossRef]
- Ahmed, A.; Badr, Y.; Shouman, S.A.; Sliem, M.A. Improvement of 6-Mercaptopurine Efficiency by Encapsulated in Chitosan Nanoparticles. Arab. J. Nucl. Sci. Appl. 2018, 51, 181–186. [Google Scholar] [CrossRef]
- Shao, X.-R.; Wei, X.-Q.; Song, X.; Hao, L.-Y.; Cai, X.-X.; Zhang, Z.-R.; Peng, Q.; Lin, Y.-F. Independent Effect of Polymeric Nanoparticle Zeta Potential/Surface Charge, on Their Cytotoxicity and Affinity to Cells. Cell Prolif. 2015, 48, 465–474. [Google Scholar] [CrossRef]
- Abdullah, M.S.P.; Noordin, M.I.; Ismail, S.I.M.; Mustapha, N.M.; Jasamai, M.; Danik, M.F.; Ismail, W.A.W.; Shamsuddin, A.F. Recent Advances in the Use of Animal-Sourced Gelatine as Natural Polymers for Food, Cosmetics and Pharmaceutical Applications. Sains Malays. 2018, 47, 323–336. [Google Scholar]
- Naomi, R.; Bahari, H.; Ridzuan, P.M.; Othman, F. Natural-Based Biomaterial for Skin Wound Healing (Gelatin vs. Collagen): Expert Review. Polymers 2021, 13, 2319. [Google Scholar] [CrossRef] [PubMed]
- Kharia, A.A.; Singhai, A.K. Effective Parameters for Formulation of Gastro Adhesive Nanoparticles: Screening by Design-of-Experiments Approach. J. Microencapsul. 2014, 31, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Kharia, A.A.; Singhai, A.K. Development and Optimisation of Mucoadhesive Nanoparticles of Acyclovir Using Design of Experiments Approach. J. Microencapsul. 2015, 32, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Gupta, Y.; Jain, A.; Saxena, A.R.; Khare, P.; Jain, A. Mannosylated Gelatin Nanoparticles Bearing an Anti-HIV Drug Didanosine for Site-Specific Delivery. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Jain, S.; Tiwary, A. Mannan-Coated Gelatin Nanoparticles for Sustained and Targeted Delivery of Didanosine: In Vitro and in Vivo Evaluation. Acta Pharm. 2008, 58, 61–74. [Google Scholar] [CrossRef]
- Tarlock, K.; Sulis, M.L.; Chewning, J.H.; Pollard, J.A.; Cooper, T.; Gamis, A.; Shenoy, S.; Kutny, M.; Horan, J.; Meshinchi, S.; et al. Hematopoietic Cell Transplantation in the Treatment of Pediatric Acute Myelogenous Leukemia and Myelodysplastic Syndromes: Guidelines from the American Society of Transplantation and Cellular Therapy. Transplant. Cell. Ther. 2022, 28, 530–545. [Google Scholar] [CrossRef]
- Ross, S.R.; McTavish, D.; Faulds, D. Fludarabine: A Review of Its Pharmacological Properties and Therapeutic Potential in Malignancy. Drugs 1993, 45, 737–759. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hargis, J.B.; Kester, K.E.; Hospenthal, D.R.; Knutson, S.W.; Diehl, L.F. Opportunistic Pulmonary Infections with Fludarabine in Previously Treated Patients with Low-Grade Lymphoid Malignancies: A Role for Pneumocystis Carinii Pneumonia Prophylaxis. Am. J. Hematol. 1995, 49, 135–142. [Google Scholar] [CrossRef]
- Sioka, C.; Kyritsis, A.P. Central and Peripheral Nervous System Toxicity of Common Chemotherapeutic Agents. Cancer Chemother. Pharmacol. 2009, 63, 761–767. [Google Scholar] [CrossRef]
- Beitinjaneh, A.; McKinney, A.M.; Cao, Q.; Weisdorf, D.J. Toxic Leukoencephalopathy Following Fludarabine-Associated Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2011, 17, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Zeman, A.; Batrakova, E.; Kabanov, A. Polyplex Nanogel Formulations for Drug Delivery of Cytotoxic Nucleoside Analogs. J. Control. Release 2005, 107, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.V.; Poluektova, L.Y.; Makarov, E.; Gerson, T.; Senanayake, M.T. Nano-NRTIs: Efficient Inhibitors of HIV Type-1 in Macrophages with a Reduced Mitochondrial Toxicity. Antivir. Chem. Chemother. 2010, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Suhail, M.; Xie, A.; Liu, J.-Y.; Hsieh, W.-C.; Lin, Y.-W.; Minhas, M.U.; Wu, P.-C. Synthesis and In Vitro Evaluation of Aspartic Acid Based Microgels for Sustained Drug Delivery. Gels 2021, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Don, T.-M.; Huang, M.-L.; Chiu, A.-C.; Kuo, K.-H.; Chiu, W.-Y.; Chiu, L.-H. Preparation of Thermo-Responsive Acrylic Hydrogels Useful for the Application in Transdermal Drug Delivery Systems. Mater. Chem. Phys. 2008, 107, 266–273. [Google Scholar] [CrossRef]
- Wang, D.; Hong, S.P.; Yang, G.; Row, K.H. Caffeine Molecular Imprinted Microgel Spheres by Precipitation Polymerization. Korean J. Chem. Eng. 2003, 20, 1073–1076. [Google Scholar] [CrossRef]
- Shaheen, S.M.; Yamaura, K. Preparation of Theophylline Hydrogels of Atactic Poly(Vinyl Alcohol)/NaCl/H2O System for Drug Delivery System. J. Control. Release 2002, 81, 367–377. [Google Scholar] [CrossRef]
- Fox, L.J.; Richardson, R.M.; Briscoe, W.H. PAMAM Dendrimer—Cell Membrane Interactions. Adv. Colloid Interface Sci. 2018, 257, 1–18. [Google Scholar] [CrossRef]
- Ziemba, B.; Matuszko, G.; Appelhans, D.; Voit, B.; Bryszewska, M.; Klajnert, B. Genotoxicity of Poly(Propylene Imine) Dendrimers. Biopolymers 2012, 97, 642–648. [Google Scholar] [CrossRef]
- Gorzkiewicz, M.; Buczkowski, A.; Appelhans, D.; Voit, B.; Pułaski, Ł.; Pałecz, B.; Klajnert-Maculewicz, B. Poly(Propyleneimine) Glycodendrimers Non-Covalently Bind ATP in a pH- and Salt-Dependent Manner—Model Studies for Adenosine Analogue Drug Delivery. Int. J. Pharm. 2018, 544, 83–90. [Google Scholar] [CrossRef]
- Gorzkiewicz, M.; Jatczak-Pawlik, I.; Studzian, M. Glycodendrimer Nanocarriers for Direct Delivery of Fludarabine Triphosphate to Leukaemic Cells: Improved Pharmacokinetics and Pharmacodynamics of Fludarabine. Biomacromolecules 2018, 19, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Gorzkiewicz, M.; Deriu, M.A.; Studzian, M.; Janaszewska, A.; Grasso, G.; Pułaski, Ł.; Appelhans, D.; Danani, A.; Klajnert-Maculewicz, B. Fludarabine-Specific Molecular Interactions with Maltose-Modified Poly(Propyleneimine) Dendrimer Enable Effective Cell Entry of the Active Drug Form: Comparison with Clofarabine. Biomacromolecules 2019, 20, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. (Ed.) Meyler’s Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions, 16th ed.; Elsevier: Amsterdam, The Netherlands; Boston, MA, USA; Heidelberg, German, 2015; ISBN 978-0-444-53716-4. [Google Scholar]
- Lotfi, K.; Månsson, E.; Spasokoukotskaja, T.; Pettersson, B.; Liliemark, J.; Peterson, C.; Eriksson, S.; Albertioni, F. Biochemical Pharmacology and Resistance to 2-Chloro-2′-Arabino-Fluoro-2′-Deoxyadenosine, a Novel Analogue of Cladribine in Human Leukemic Cells. Clin. Cancer Res. 1999, 5, 2438–2444. [Google Scholar] [PubMed]
- Ghanem, H.; Kantarjian, H.; Ohanian, M.; Jabbour, E. The Role of Clofarabine in Acute Myeloid Leukemia. Leuk. Lymphoma 2013, 54, 688–698. [Google Scholar] [CrossRef]
- Fozza, C. The Role of Clofarabine in the Treatment of Adults with Acute Myeloid Leukemia. Crit. Rev. Oncol. Hematol. 2015, 93, 237–245. [Google Scholar] [CrossRef]
- Jeha, S.; Razzouk, B.; Rytting, M.; Rheingold, S.; Albano, E.; Kadota, R.; Luchtman-Jones, L.; Bomgaars, L.; Gaynon, P.; Goldman, S.; et al. Phase II Study of Clofarabine in Pediatric Patients with Refractory or Relapsed Acute Myeloid Leukemia. JCO 2009, 27, 4392–4397. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Xiao, L.; Garcia-Manero, G.; Plunkett, W.; Gandhi, V.; Sasaki, K.; Pemmaraju, N.; et al. A Randomized Phase 2 Study of Idarubicin and Cytarabine with Clofarabine or Fludarabine in Patients with Newly Diagnosed Acute Myeloid Leukemia: CIA Versus FIA in AML. Cancer 2017, 123, 4430–4439. [Google Scholar] [CrossRef]
- Neerman, M.F.; Chen, H.-T.; Parrish, A.R.; Simanek, E.E. Reduction of Drug Toxicity Using Dendrimers Based on Melamine. Mol. Pharm. 2004, 1, 390–393. [Google Scholar] [CrossRef]
- Neerman, M.F. The Efficiency of a PAMAM Dendrimer toward the Encapsulation of the Antileukemic Drug 6-Mercaptopurine. Anti-Cancer Drugs 2007, 18, 839–842. [Google Scholar] [CrossRef]
- Hübener, S.; Oo, Y.H.; Than, N.N.; Hübener, P.; Weiler-Normann, C.; Lohse, A.W.; Schramm, C. Efficacy of 6-Mercaptopurine as Second-Line Treatment for Patients With Autoimmune Hepatitis and Azathioprine Intolerance. Clin. Gastroenterol. Hepatol. 2016, 14, 445–453. [Google Scholar] [CrossRef]
- Wang, X.; Cai, X.; Hu, J.; Shao, N.; Wang, F.; Zhang, Q.; Xiao, J.; Cheng, Y. Glutathione-Triggered “Off–On” Release of Anticancer Drugs from Dendrimer-Encapsulated Gold Nanoparticles. J. Am. Chem. Soc. 2013, 135, 9805–9810. [Google Scholar] [CrossRef] [PubMed]
- Yandrapu, S.K.; Kanujia, P.; Chalasani, K.B.; Mangamoori, L.; Kolapalli, R.V.; Chauhan, A. Development and Optimization of Thiolated Dendrimer as a Viable Mucoadhesive Excipient for the Controlled Drug Delivery: An Acyclovir Model Formulation. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, F.; Huang, J.; Lu, W.; Liu, S.; Yu, J. Fabrication of Biodegradable Micelles with Reduction-Triggered Release of 6-Mercaptopurine Profile Based on Disulfide-Linked Graft Copolymer Conjugate. Colloids Surf. B Biointerfaces 2012, 100, 155–162. [Google Scholar] [CrossRef]
- Shi, L.; Ding, K.; Sun, X.; Zhang, L.; Zeng, T.; Yin, Y.; Zheng, H. Preparation, Characterization, and in Vitro Drug Release Behavior of Glutathione-Sensitive Long-Circulation Micelles Based on Polyethylene Glycol Prodrug. J. Biomater. Sci. Polym. Ed. 2016, 27, 472–489. [Google Scholar] [CrossRef]
- Liao, J.; Peng, H.; Wei, X.; Song, Y.; Liu, C.; Li, D.; Yin, Y.; Xiong, X.; Zheng, H.; Wang, Q. A Bio-Responsive 6-Mercaptopurine/Doxorubicin Based “Click Chemistry” Polymeric Prodrug for Cancer Therapy. Mater. Sci. Eng. C 2020, 108, 110461. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Hearn, M.T.W.; Bell, T.D.M.; Saito, K. Release Kinetics of 6-Mercaptopurine and 6-Thioguanine from Bioinspired Core-Crosslinked Thymine Functionalised Polymeric Micelles. Aust. J. Chem. 2013, 66, 952. [Google Scholar] [CrossRef]
- Jeanbart, L.; Kourtis, I.C.; van der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. 6-Thioguanine-Loaded Polymeric Micelles Deplete Myeloid-Derived Suppressor Cells and Enhance the Efficacy of T Cell Immunotherapy in Tumor-Bearing Mice. Cancer Immunol. Immunother. 2015, 64, 1033–1046. [Google Scholar] [CrossRef]
- Zheng, H.; Rao, Y.; Yin, Y.; Xiong, X.; Xu, P.; Lu, B. Preparation, Characterization, and in Vitro Drug Release Behavior of 6-Mercaptopurine-Carboxymethyl Chitosan. Carbohydr. Polym. 2011, 83, 1952–1958. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The Role of Glutathione in Cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Sawdon, A.J.; Peng, C.-A. Polymeric Micelles for Acyclovir Drug Delivery. Colloids Surf. B Biointerfaces 2014, 122, 738–745. [Google Scholar] [CrossRef]
- Sawdon, A.; Peng, C.-A. Ring-Opening Polymerization of ε-Caprolactone Initiated by Ganciclovir (GCV) for the Preparation of GCV-Tagged Polymeric Micelles. Molecules 2015, 20, 2857–2867. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Cmax (µg/mL) | AUC0–∞ (μg/mL·h) | t1/2 (h) | Clearance (L/h) | MRT (h) |
|---|---|---|---|---|---|
| Commercial | 26.37 | 38.52 | 1.34 | 0.5284 | 2.11 |
| PLGA-ACV | 23.15 | 803.34 | 45.10 | 0.0252 | 63.22 |
| API | Ø (nm) | ζ (mV) | EE (%) | Summary | Ref. |
|---|---|---|---|---|---|
| 6-MP | 138.01 | −1.0 | 80.71 |
| [46] |
| 6-TG | 149.10 | −36.6 | 97.22 |
| [47] |
| ACV | 285 | −10.1 | 14.02 |
| [58] |
| 198.1 | −8.5 | 61.10 |
| [9] | |
| 262 | 15.14 | 74.12 |
| [7] | |
| 164.9 | n.g. | 59.7 |
| [8] | |
| 740 | n.g. | 54.4 |
| [60] | |
| 400 | 28.73 | 80.16 |
| [63] | |
| 161 | −6.7 | n.g. |
| [10] | |
| TNF | 275 | −29.5 | n.g. |
| [73] |
| 127 | 48.4 | n.g. |
| [74] | |
| 300–700 | >0 | n.g. |
| [76] | |
| TDF | 336.8 | −2.38 | 21.9 |
| [72] |
| 148.6 | −26.7 | 24 |
| [75] | |
| 218 | −4.8 | 52.9 |
| [77] | |
| To sum up | |||||
| Pros | |||||
| |||||
| Cons | |||||
| |||||
| API | Ø (nm) | ζ (mV) | EE (%) | Summary | Ref. |
|---|---|---|---|---|---|
| ACV | 236 | n.g. | 79.34 |
| [20] |
| 99 | 26.1 | n.g. |
| [84] |
| Gelatin ↑ | Stabilizer ↑ | Cross-Linking Agent ↑ | Acetone ↑ | pH ↑ | Stirring Speed ↑ | Stirring Time ↑ | |
|---|---|---|---|---|---|---|---|
| Size | n.g. | ↓ | ↓ | n.g. | ↑ | ↓ | n.g. |
| PDI | ↓ | n.g. | ↑ | n.g. | ↑* | ↓ | n.g. |
| Loading efficiency | n.g. | ↑ | n.g. | ↑ | * | ↓ | ↑ |
| Drug release | ↓ | ↓ | n.g. | n.g. | n.g. | n.g. | n.g. |
| Mucoadhesion | ↑ | ↓ | ↓ | n.g | n.g | n.g | n.g |
| API | Material | Ø (nm) | ζ (mV) | EE (%) | Summary | Ref. |
|---|---|---|---|---|---|---|
| ACV | Gelatin | 165–1610 | n.g. | 39–80 |
| [111] |
| Gelatin | 274.4 | n.g. | 70.65 |
| [112] | |
| DDI | Gelatin | 325 | 6.2 | n.a. |
| [113] |
| Gelatin | 140 | 7.2 | 79.5 |
| [114] | |
| PEI | 90–120 | n.g. | n.g. |
| [121] | |
| FLU | PEI | 58 | n.g. | 130 µg/mg |
| [120] |
| To sum up | ||||||
| Pros | ||||||
| ||||||
| Cons | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szyk, P.; Czarczynska-Goslinska, B.; Mlynarczyk, D.T.; Ślusarska, B.; Kocki, T.; Ziegler-Borowska, M.; Goslinski, T. Polymer-Based Nanoparticles as Drug Delivery Systems for Purines of Established Importance in Medicine. Nanomaterials 2023, 13, 2647. https://doi.org/10.3390/nano13192647
Szyk P, Czarczynska-Goslinska B, Mlynarczyk DT, Ślusarska B, Kocki T, Ziegler-Borowska M, Goslinski T. Polymer-Based Nanoparticles as Drug Delivery Systems for Purines of Established Importance in Medicine. Nanomaterials. 2023; 13(19):2647. https://doi.org/10.3390/nano13192647
Chicago/Turabian StyleSzyk, Piotr, Beata Czarczynska-Goslinska, Dariusz T. Mlynarczyk, Barbara Ślusarska, Tomasz Kocki, Marta Ziegler-Borowska, and Tomasz Goslinski. 2023. "Polymer-Based Nanoparticles as Drug Delivery Systems for Purines of Established Importance in Medicine" Nanomaterials 13, no. 19: 2647. https://doi.org/10.3390/nano13192647