

Mechanical Exfoliation of Expanded Graphite to Graphene-Based Materials and Modification with Palladium Nanoparticles for Hydrogen Storage
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of EG
2.3. Synthesis of Mechanically Exfoliated Nanomaterials
2.4. Synthesis of ShEG Nanomaterials
2.5. Synthesis of sEG Nanomaterials
2.6. Synthesis of Pd-Decorated Nanocomposites
2.7. Structural Characterization
2.8. Electrochemical Measurements
3. Results and Discussion
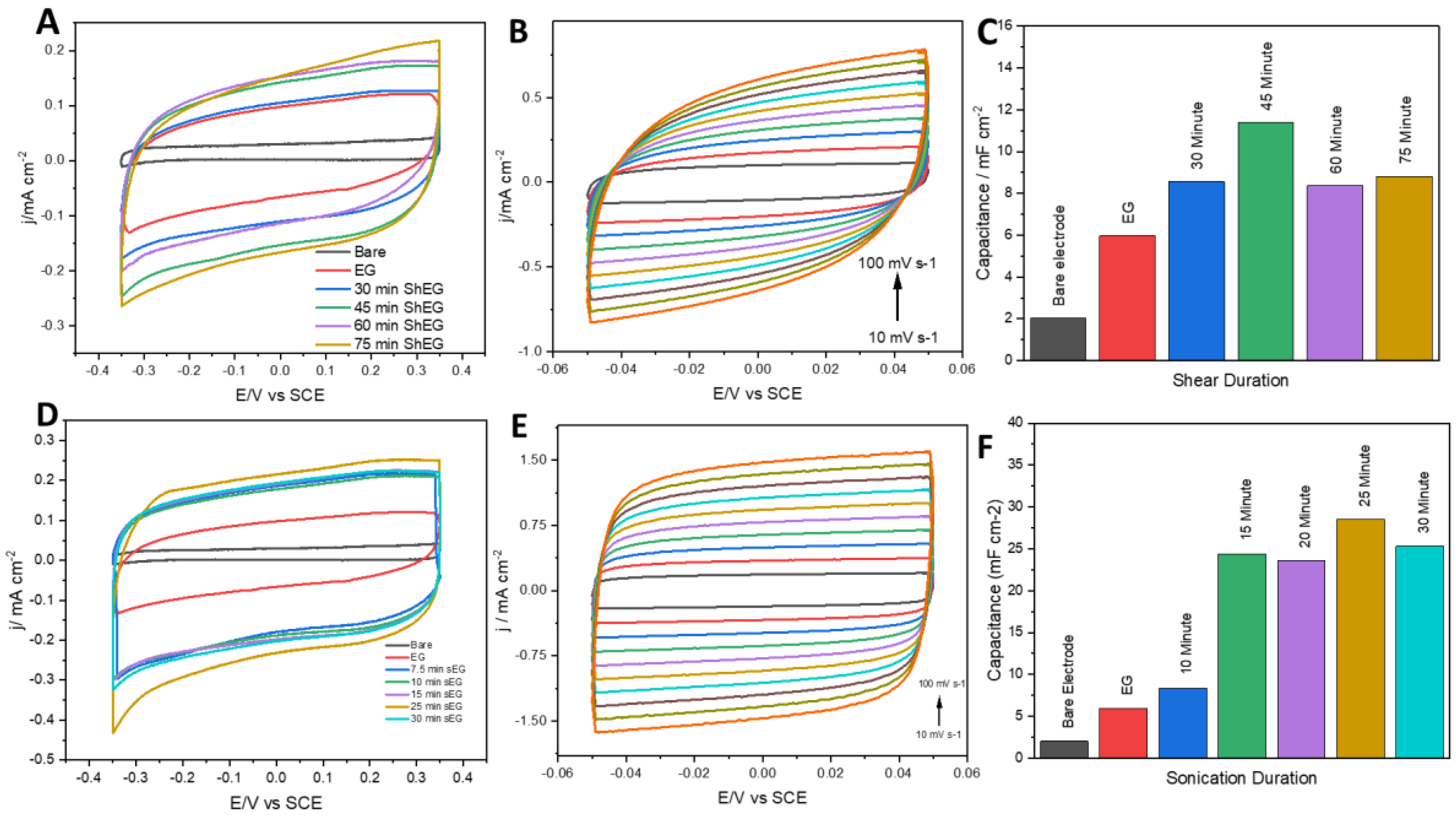
3.1. Capacitance Studies of Mechanically Exfoliated EG
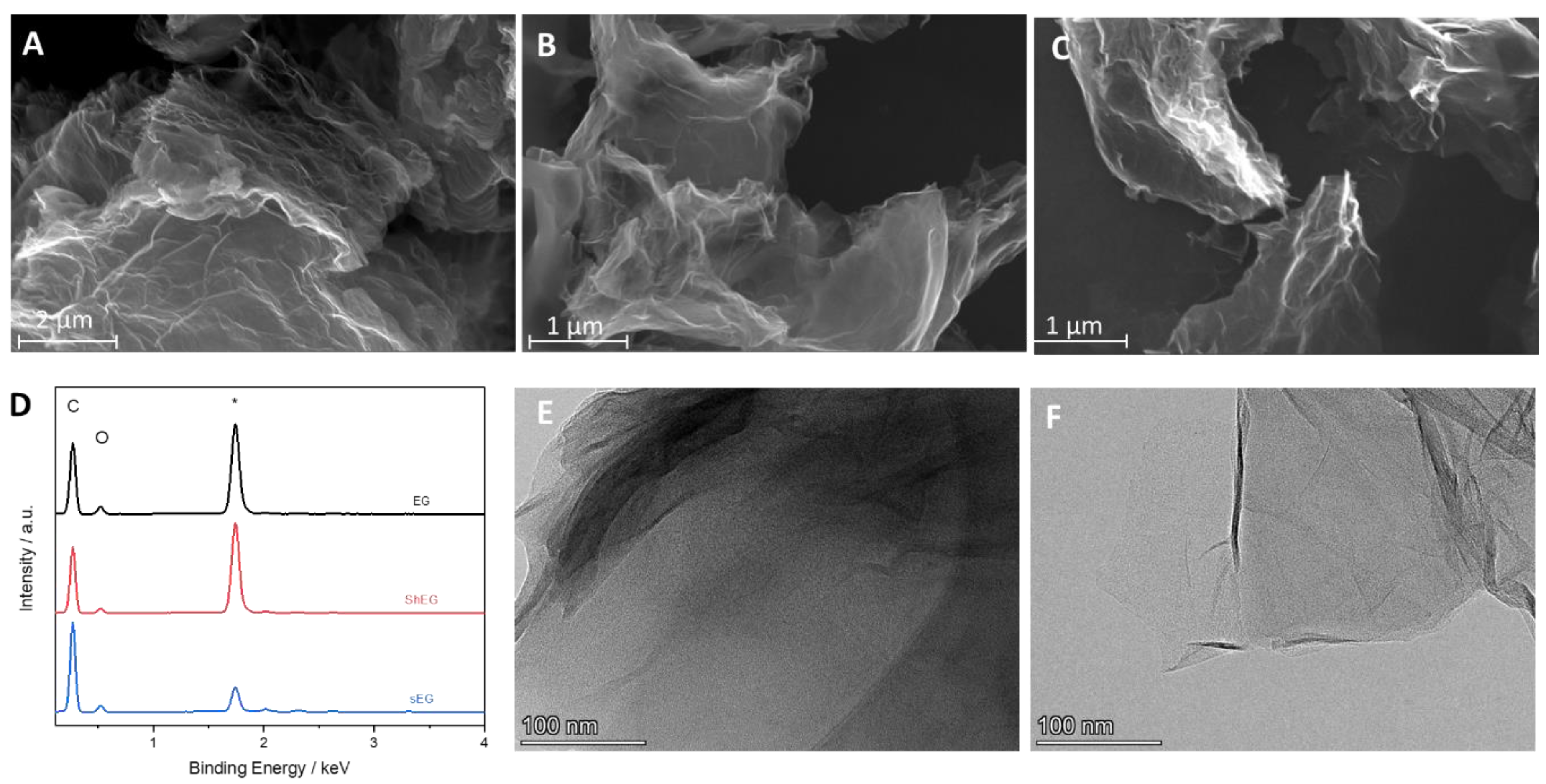
3.2. Morphological Characterization of Graphene-Based Nanomaterials
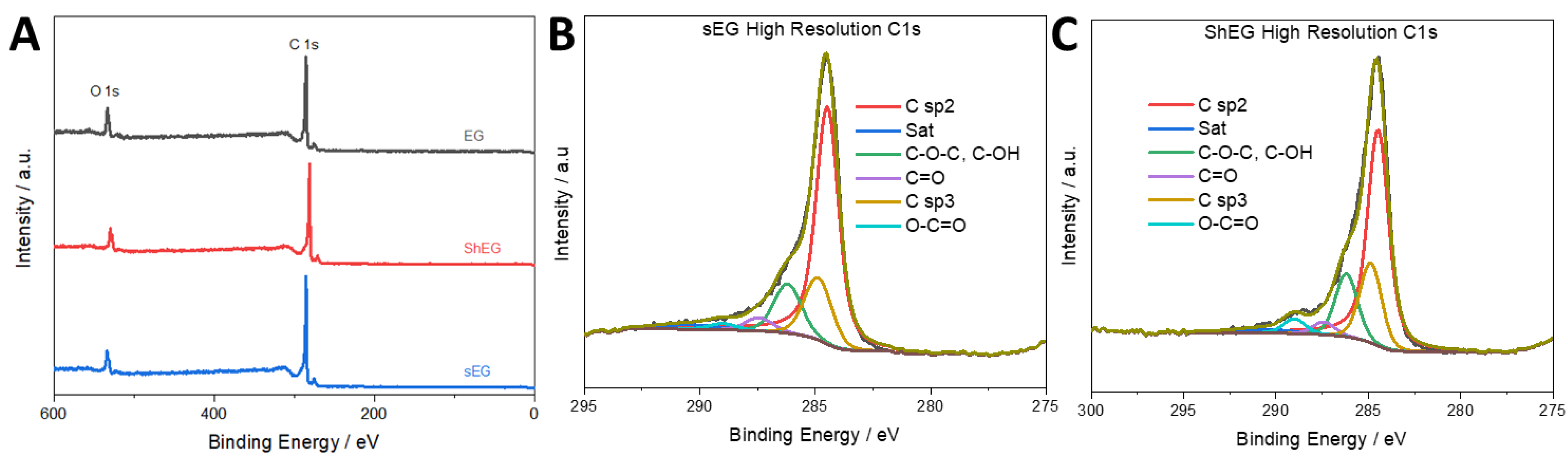
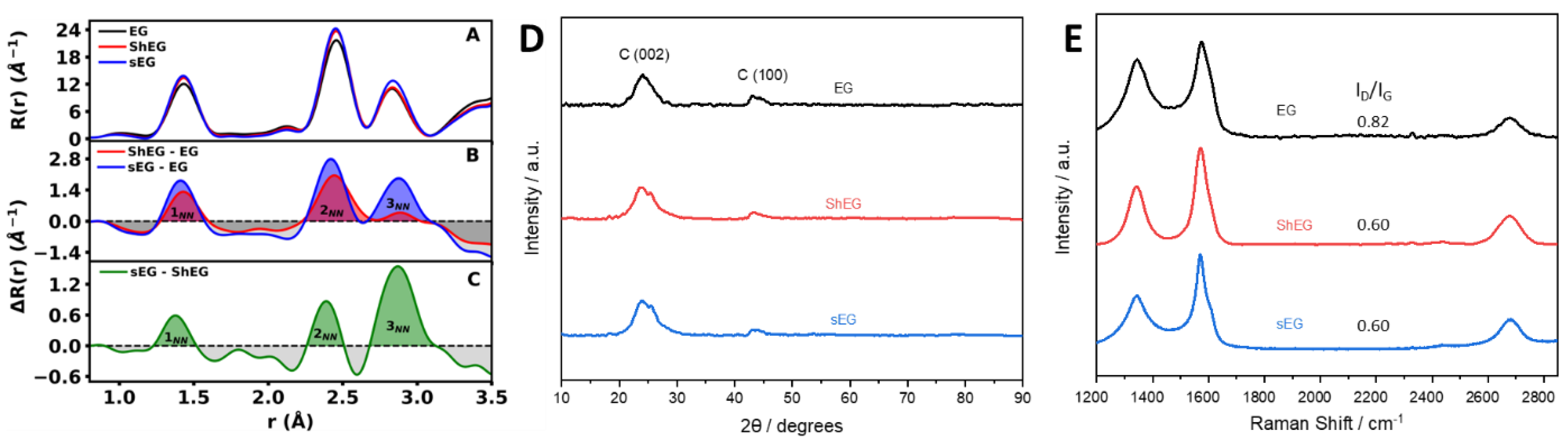
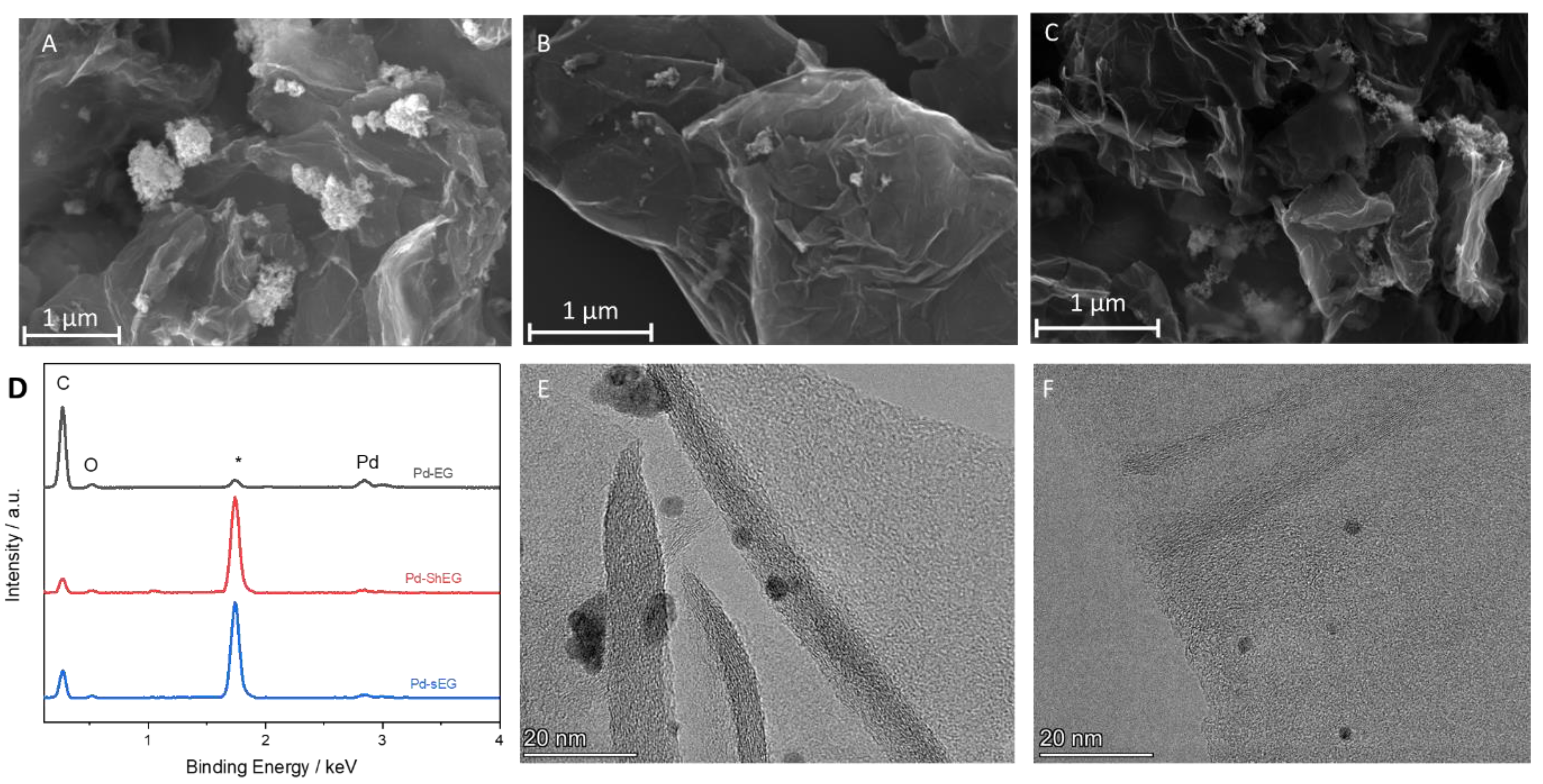
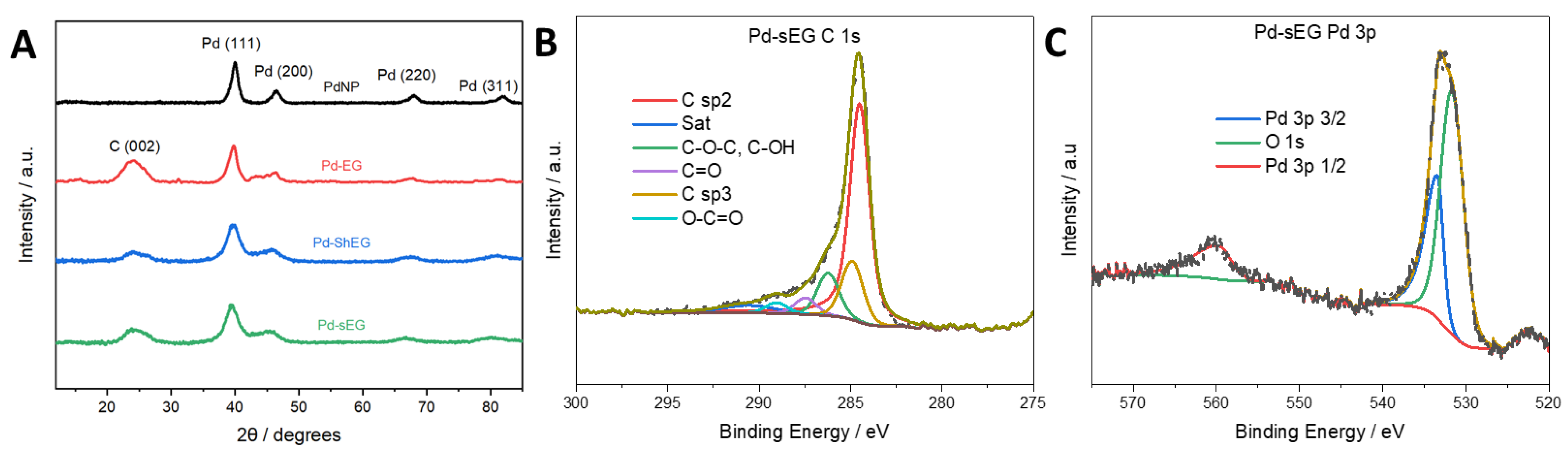
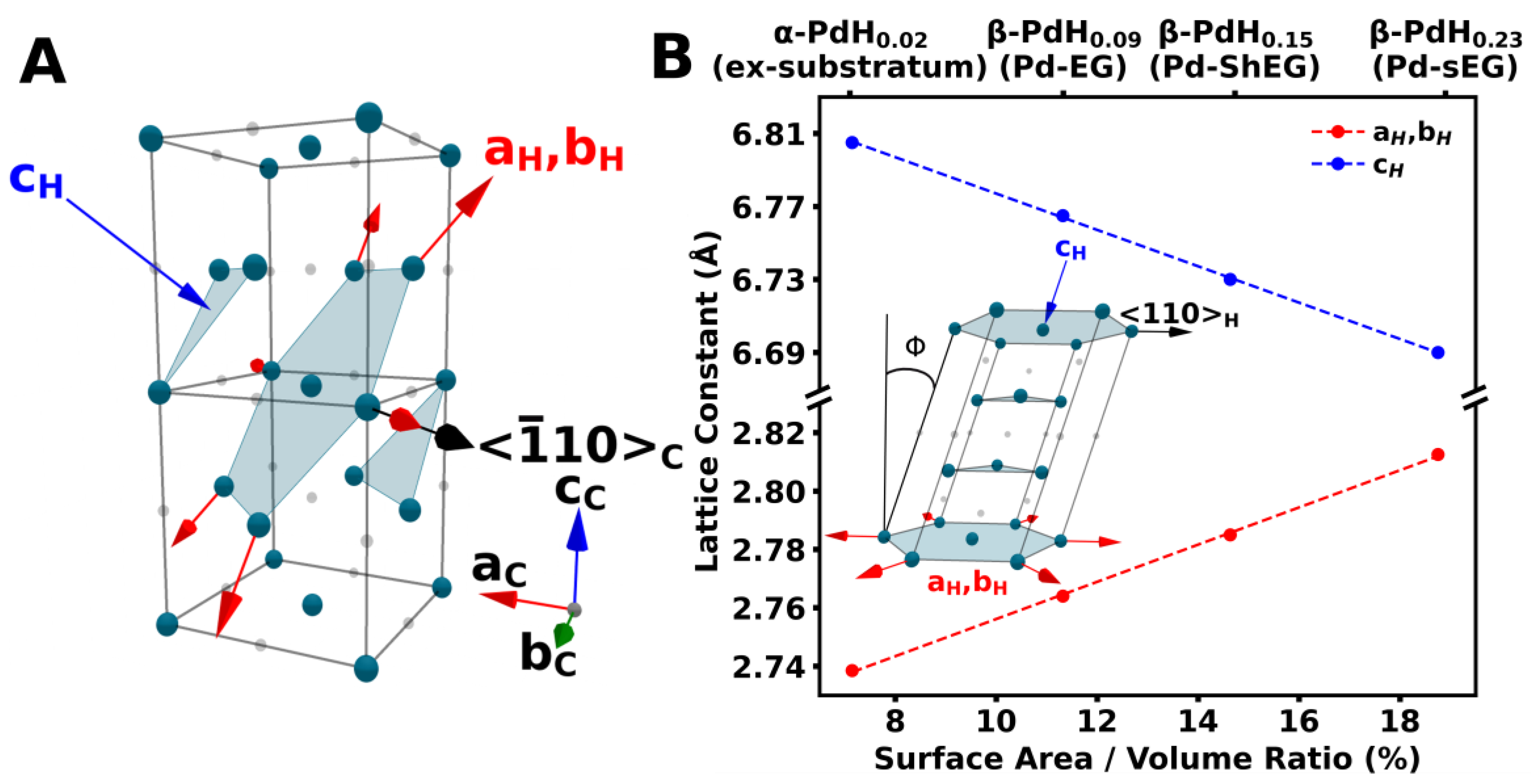
3.3. Characterization of Palladium Nanoparticle Functionalized Nanocomposites
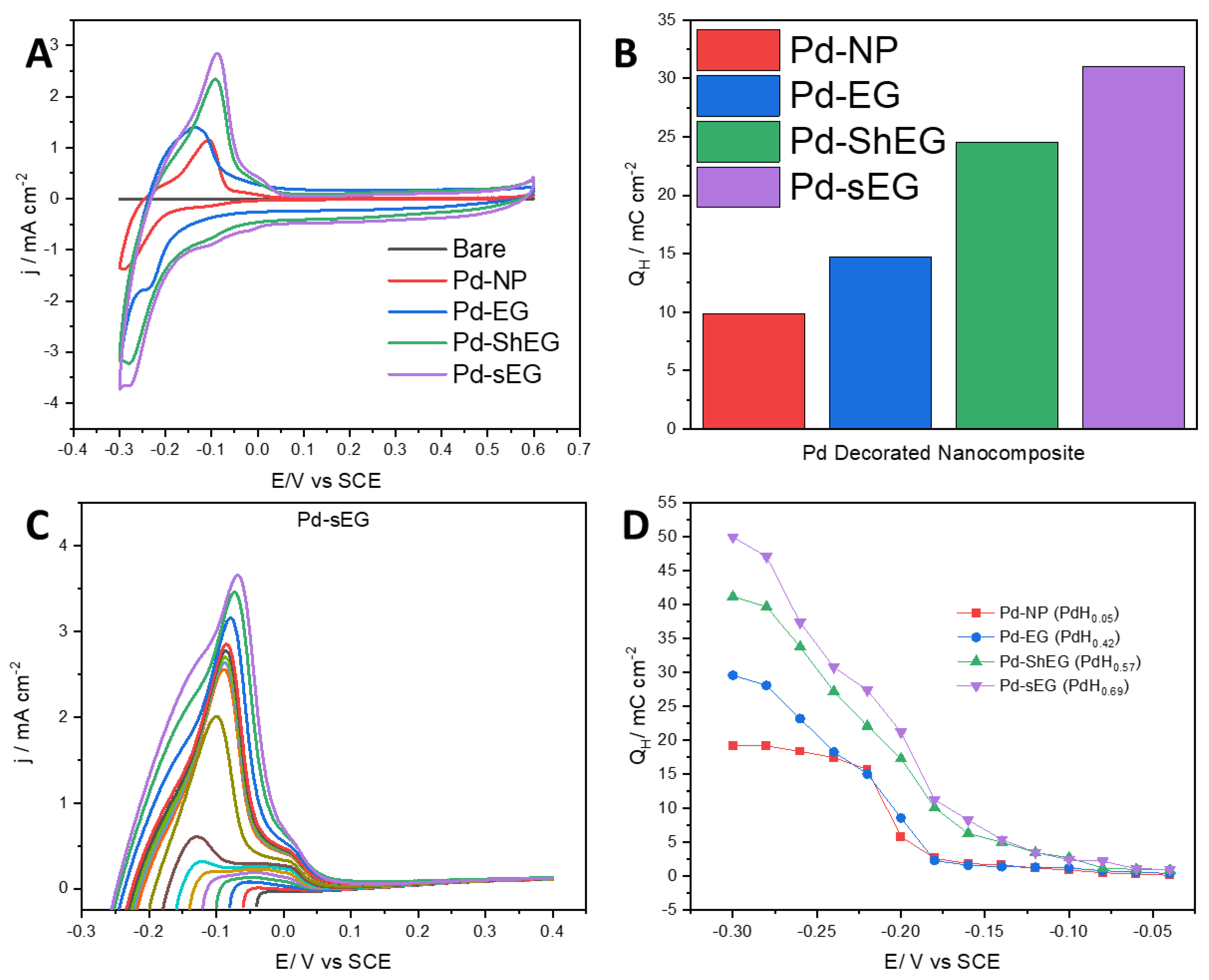
3.4. Hydrogen Uptake and Release Performance of Pd-EG Nanomaterials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crabtree, G.W.; Dresselhaus, M.S. The Hydrogen Fuel Alternative. MRS Bull. 2008, 33, 421–428. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-Storage Materials for Mobile Applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, N.; Alcalde, J.; Miocic, J.M.; Hangx, S.J.T.; Kallmeyer, J.; Ostertag-Henning, C.; Hassanpouryouzband, A.; Thaysen, E.M.; Strobel, G.J.; Schmidt-Hattenberger, C.; et al. Enabling Large-Scale Hydrogen Storage in Porous Media-the Scientific Challenges. Energy Environ. Sci. 2021, 14, 853–864. [Google Scholar] [CrossRef]
- Tzimas, E.; Filiou, C.; Peteves, S.D.; Veyret, J. Hydrogen Storage: State-of-the-Art and Future Perspective; European Commission: Petten, The Netherlands, 2003; ISBN 92-894-6950-1. [Google Scholar]
- Boateng, E.; Chen, A. Recent Advances in Nanomaterial-Based Solid-State Hydrogen Storage. Mater. Today Adv. 2020, 6, 100022. [Google Scholar] [CrossRef]
- Boateng, E.; Thiruppathi, A.R.; Hung, C.K.; Chow, D.; Sridhar, D.; Chen, A. Functionalization of Graphene-Based Nanomaterials for Energy and Hydrogen Storage. Electrochim. Acta 2023, 452, 142340. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, B.; Zhang, L.; Zhu, Y.; Gong, W. Monolayer AsC5 as the Promising Hydrogen Storage Material for Clean Energy Applications. Nanomaterials 2023, 13, 1553. [Google Scholar] [CrossRef] [PubMed]
- Pal, N.; Chakraborty, D.; Cho, E.-B.; Seo, J.G. Recent Developments on the Catalytic and Biosensing Applications of Porous Nanomaterials. Nanomaterials 2023, 13, 2184. [Google Scholar] [CrossRef]
- Wei, L.; Mao, Y. Enhanced Hydrogen Storage Performance of Reduced Graphene Oxide Hybrids with Nickel or Its Metallic Mixtures Based on Spillover Mechanism. Int. J. Hydrogen Energy 2016, 41, 11692–11699. [Google Scholar] [CrossRef]
- Huo, Y.; Zhang, Y.; Wang, C.; Fang, Y.; Li, K.; Chen, Y. Boron-Doping Effect on the Enhanced Hydrogen Storage of Titanium-Decorated Porous Graphene: A First-Principles Study. Int. J. Hydrogen Energy 2021, 46, 40301–40311. [Google Scholar] [CrossRef]
- Wang, P.; Xia, K.; Chen, Y.; Tian, Q.; Xiong, R.; Han, B.; Gao, Q.; Zhou, C.; Yu, D. Acid-Assisted Synthesis of Nitrogen-Deficient Mesoporous Graphitic Carbon Nitride for Hydrogen Storage. Mater. Lett. 2021, 301, 130347. [Google Scholar] [CrossRef]
- Mosquera-Vargas, E.; Tamayo, R.; Morel, M.; Roble, M.; Díaz-Droguett, D.E. Hydrogen Storage in Purified Multi-Walled Carbon Nanotubes: Gas Hydrogenation Cycles Effect on the Adsorption Kinetics and Their Performance. Heliyon 2021, 7, e08494. [Google Scholar] [CrossRef] [PubMed]
- Lobo, R.; Ribeiro, J.; Inok, F. Hydrogen Uptake and Release in Carbon Nanotube Electrocatalysts. Nanomaterials 2021, 11, 975. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.; Ojha, R.; Rezaei Niya, S.M.; Seibt, S. Electrochemical Storage Reactions of Hydrogen in Activated Carbon from Phenolic Resin. Catal. Today 2021, 397–399, 155–164. [Google Scholar] [CrossRef]
- Thiruppathi, A.R.; Sidhureddy, B.; Boateng, E.; Soldatov, D.V.; Chen, A. Synthesis and Electrochemical Study of Three-Dimensional Graphene-Based Nanomaterials for Energy Applications. Nanomaterials 2020, 10, 1295. [Google Scholar] [CrossRef]
- Adams, B.D.; Ostrom, C.K.; Chen, S.; Chen, A. High-Performance Pd-Based Hydrogen Spillover Catalysts for Hydrogen Storage. J. Phys. Chem. C 2010, 114, 19875–19882. [Google Scholar] [CrossRef]
- Parambhath, V.B.; Nagar, R.; Sethupathi, K.; Ramaprabhu, S. Investigation of Spillover Mechanism in Palladium Decorated Hydrogen Exfoliated Functionalized Graphene. J. Phys. Chem. C 2011, 115, 15679–15685. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, Y.; Wang, Y.; Liu, T.; Cui, H. A Study on the Hydrogen Storage Performance of Graphene–Pd(T)–Graphene Structure. Int. J. Hydrogen Energy 2020, 45, 12376–12383. [Google Scholar] [CrossRef]
- Boateng, E.; Van Der Zalm, J.; Chen, A. Design and Electrochemical Study of Three-Dimensional Expanded Graphite and Reduced Graphene Oxide Nanocomposites Decorated with Pd Nanoparticles for Hydrogen Storage. J. Phys. Chem. C 2021, 125, 22970–22981. [Google Scholar] [CrossRef]
- Paton, K.R.; Varrla, E.; Backes, C.; Smith, R.J.; Khan, U.; O’Neill, A.; Boland, C.; Lotya, M.; Istrate, O.M.; King, P.; et al. Scalable Production of Large Quantities of Defect-Free Few-Layer Graphene by Shear Exfoliation in Liquids. Nat. Mater. 2014, 13, 624–630. [Google Scholar] [CrossRef]
- Lynch-Branzoi, J.K.; Ashraf, A.; Tewatia, A.; Taghon, M.; Wooding, J.; Hendrix, J.; Kear, B.H.; Nosker, T.J. Shear Exfoliation of Graphite into Graphene Nanoflakes Directly within Polyetheretherketone and a Spectroscopic Study of This High Modulus, Lightweight Nanocomposite. Compos. B Eng. 2020, 188, 107842. [Google Scholar] [CrossRef]
- Liang, S.; Shen, Z.; Yi, M.; Liu, L.; Zhang, X.; Ma, S. In-Situ Exfoliated Graphene for High-Performance Water-Based Lubricants. Carbon N. Y. 2016, 96, 1181–1190. [Google Scholar] [CrossRef]
- Liang, B.; Liu, K.; Liu, P.; Qian, L.; Zhao, G.; Pan, W.; Chen, C. Organic Salt-Assisted Liquid-Phase Shear Exfoliation of Expanded Graphite into Graphene Nanosheets. J. Mater. 2021, 7, 1181–1189. [Google Scholar] [CrossRef]
- Costinas, C.; Salagean, C.A.; Cotet, L.C.; Baia, M.; Todea, M.; Magyari, K.; Baia, L. Insights into the Stability of Graphene Oxide Aqueous Dispersions. Nanomaterials 2022, 12, 4489. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Lund, S.; Kauppila, J.; Sirkiä, S.; Palosaari, J.; Eklund, O.; Latonen, R.M.; Smått, J.H.; Peltonen, J.; Lindfors, T. Fast High-Shear Exfoliation of Natural Flake Graphite with Temperature Control and High Yield. Carbon N. Y. 2021, 174, 123–131. [Google Scholar] [CrossRef]
- Gao, M.; Zong, H.; Li, Y.; Zhou, Y.; Yu, L.; Qin, L.; Zhao, S. Novel Cyclic Ultrasound-Assisted Liquid Phase Exfoliation of Graphene in Deionized Water: A Parameter Study. Mater. Lett. 2023, 337, 134011. [Google Scholar] [CrossRef]
- Tyurnina, A.V.; Morton, J.A.; Kaur, A.; Mi, J.; Grobert, N.; Porfyrakis, K.; Tzanakis, I.; Eskin, D.G. Effects of Green Solvents and Surfactants on the Characteristics of Few-Layer Graphene Produced by Dual-Frequency Ultrasonic Liquid Phase Exfoliation Technique. Carbon N. Y. 2023, 206, 7–15. [Google Scholar] [CrossRef]
- Sidhureddy, B.; Thiruppathi, A.R.; Chen, A. From Graphite to Interconnected Reduced Graphene Oxide: One-Pot Synthesis and Supercapacitor Application. Chem. Commun. 2017, 53, 7828–7831. [Google Scholar] [CrossRef]
- Liu, L.; Qing, M.; Wang, Y.; Chen, S. Defects in Graphene: Generation, Healing, and Their Effects on the Properties of Graphene: A Review. J. Mater. Sci. Technol. 2015, 31, 599–606. [Google Scholar] [CrossRef]
- Alshamkhani, M.T.; Keat, L.; Kurnianditia, L.; Rahman, A.; Lahijani, P.; Mohammadi, M. Journal of Environmental Chemical Engineering Effect of Graphite Exfoliation Routes on the Properties of Exfoliated Graphene and Its Photocatalytic Applications. J. Environ. Chem. Eng. 2021, 9, 106506. [Google Scholar] [CrossRef]
- Huang, H.; Wang, X. Pd Nanoparticles Supported on Low-Defect Graphene Sheets: For Use as High-Performance Electrocatalysts for Formic Acid and Methanol Oxidation. J. Mater. Chem. 2012, 22, 22533–22541. [Google Scholar] [CrossRef]
- Baird, A.; Andrews, J. Storage of Atomic Hydrogen in Multilayer Graphene. Int. J. Hydrogen Energy 2023, 48, 27944–27959. [Google Scholar] [CrossRef]
- Li, Z.; Gadipelli, S.; Yang, Y.; Guo, Z. Design of 3D Graphene-Oxide Spheres and Their Derived Hierarchical Porous Structures for High Performance Supercapacitors. Small 2017, 13, 1702474. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Ho, H.N.; Hsu, K.T.; Liu, W.R. Improvement of Electromagnetic Interference Properties of 3D Few-Layer Graphene Composite by Means of Freeze-Drying. Ceram Int. 2022, 48, 26107–26115. [Google Scholar] [CrossRef]
- Gomez, A.; Dina, G.; Kycia, S. The High-Energy X-ray Diffraction and Scattering Beamline at the Canadian Light Source. Rev. Sci. Instrum. 2018, 89, 063301. [Google Scholar] [CrossRef]
- Burns, N.; Rahemtulla, A.; Annett, S.; Moreno, B.; Kycia, S. An Inclined Detector Geometry for Improved X-ray Total Scattering Measurements. J. Appl. Crystallogr. 2023, 56, 510–518. [Google Scholar] [CrossRef]
- Farrow, C.L.; Juhas, P.; Liu, J.W.; Bryndin, D.; Boin, E.S.; Bloch, J.; Proffen, T.; Billinge, S.J.L. PDFfit2 and PDFgui: Computer Programs for Studying Nanostructure in Crystals. J. Phys. Condens. Matter. 2007, 19, 335219. [Google Scholar] [CrossRef]
- Li, Z.; He, W.; Wang, X.; Wang, X.; Song, M.; Zhao, J. N/S Dual-Doped Graphene with High Defect Density for Enhanced Supercapacitor Properties. Int. J. Hydrogen Energy 2020, 45, 112–122. [Google Scholar] [CrossRef]
- Hulman, M. Raman Spectroscopy of Graphene. In Graphene: Properties, Preparation, Characterization and Applications, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 381–411. ISBN 9780081028483. [Google Scholar]
- Stevie, F.A.; Donley, C.L. Introduction to X-ray Photoelectron Spectroscopy. J. Vac. Sci. Technol. A 2020, 38, 063204. [Google Scholar] [CrossRef]
- Morelhão, S.L.; Kycia, S.W. A simple formula for determining nanoparticle size distribution by combining small-angle X-ray scattering and diffraction results. Acta Crystallogr. Sect. A Found. Adv. 2022, 78, 1. [Google Scholar] [CrossRef]
- Pinos-Vélez, V.; Osegueda, O.; Crivoi, D.G.; Llorca, J.; García-García, F.J.; Álvarez, M.G.; Medina, F.; Dafinov, A. Insights into Palladium Deactivation during Advanced Oxidation Processes. Chem. Mater. 2022, 34, 8760–8768. [Google Scholar] [CrossRef] [PubMed]
- Bugaev, A.L.; Guda, A.A.; Lomachenko, K.A.; Shapovalov, V.V.; Lazzarini, A.; Vitillo, J.G.; Bugaev, L.A.; Groppo, E.; Pellegrini, R.; Soldatov, A.V.; et al. Core-Shell Structure of Palladium Hydride Nanoparticles Revealed by Combined X-ray Absorption Spectroscopy and X-ray Diffraction. J. Phys. Chem. C 2017, 121, 18202–18213. [Google Scholar] [CrossRef]
- Akiba, H.; Kofu, M.; Kobayashi, H.; Kitagawa, H.; Ikeda, K.; Otomo, T.; Yamamuro, O. Nanometer-Size Effect on Hydrogen Sites in Palladium Lattice. J. Am. Chem. Soc. 2016, 138, 10238–10243. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Zhao, C.; Paterson, A.R.; Li, S.; Jones, J.L. Local Structures of Perovskite Dielectrics and Ferroelectrics via Pair Distribution Function Analyses. J. Eur. Ceram Soc. 2018, 38, 971–987. [Google Scholar] [CrossRef]
- Kennedy, B.J.; Vogt, T. Powder X-ray Diffraction Study of the Rhombohedral to Cubic Phase Transition in TiF 3. Res. Bull. 2002, 37, 77–83. [Google Scholar] [CrossRef]
- Ma, Z.; Tan, L.; Huang, H.; He, L.; Chen, J.; Lu, H.; Deng, S.; Yin, W.; Zhang, J.; Tian, H.; et al. Neutron Powder-Diffraction Study of Phase Transitions in Strontium-Doped Bismuth Ferrite: 1. Variation with Chemical Composition. Phys. Condens. Matter. 2022, 34, 255401. [Google Scholar] [CrossRef]
- Zheng, Z.; Su, X.; Deng, R.; Stoumpos, C.; Xie, H.; Liu, W.; Yan, Y.; Hao, S.; Uher, C.; Wolverton, C.; et al. Rhombohedral to Cubic Conversion of GeTe via MnTe Alloying Leads to Ultralow Thermal Conductivity, Electronic Band Convergence, and High Thermoelectric Performance. J. Am. Chem. Soc. 2018, 140, 2673–2686. [Google Scholar] [CrossRef]
- Schirber, J.E.; Morosin, B. Lattice Constants of β-PdHx and P-PdDx with x near 1.0. Phys. Rev. B 1975, 12, 117. [Google Scholar] [CrossRef]
- Hijazi, I.; Zhang, Y.; Fuller, R. A Simple Palladium Hydride Embedded Atom Method Potential for Hydrogen Energy Applications. J. Energy Resour. Technol. Trans. ASME 2019, 141, 061202. [Google Scholar] [CrossRef]
- Zhou, X.W.; Zimmerman, J.A.; Wong, B.M.; Hoyt, J.J. An Embedded-Atom Method Interatomic Potential for Pd-H Alloys. J. Mater. Res. 2008, 23, 704–718. [Google Scholar] [CrossRef]
- Askari, M.B.; Salarizadeh, P. Ultra-Small ReS2 Nanoparticles Hybridized with RGO as Cathode and Anode Catalysts towards Hydrogen Evolution Reaction and Methanol Electro-Oxidation for DMFC in Acidic and Alkaline Media. Synth. Met. 2019, 256, 116131. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Applicability of Platinum as a Counter-Electrode Material in Electrocatalysis Research. ACS Catal. 2022, 12, 2661–2670. [Google Scholar] [CrossRef]
- Wheeler, J.M.; Clyne, T.W. Nanoindentation of Palladium-Hydrogen. Int. J. Hydrogen Energy 2012, 37, 14315–14322. [Google Scholar] [CrossRef]
- Manchester, F.D.; San-Martin, A.; Pitre, J.M. The H-Pd (Hydrogen-Palladium) System. J. Phase Equilibria 1994, 15, 62–83. [Google Scholar] [CrossRef]
- Chen, S.; Adams, B.D.; Chen, A. Synthesis and Electrochemical Study of Nanoporous Pd-Ag Alloys for Hydrogen Sorption. Electrochim. Acta 2010, 56, 61–67. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chow, D.; Burns, N.; Boateng, E.; van der Zalm, J.; Kycia, S.; Chen, A. Mechanical Exfoliation of Expanded Graphite to Graphene-Based Materials and Modification with Palladium Nanoparticles for Hydrogen Storage. Nanomaterials 2023, 13, 2588. https://doi.org/10.3390/nano13182588
Chow D, Burns N, Boateng E, van der Zalm J, Kycia S, Chen A. Mechanical Exfoliation of Expanded Graphite to Graphene-Based Materials and Modification with Palladium Nanoparticles for Hydrogen Storage. Nanomaterials. 2023; 13(18):2588. https://doi.org/10.3390/nano13182588
Chicago/Turabian StyleChow, Darren, Nicholas Burns, Emmanuel Boateng, Joshua van der Zalm, Stefan Kycia, and Aicheng Chen. 2023. "Mechanical Exfoliation of Expanded Graphite to Graphene-Based Materials and Modification with Palladium Nanoparticles for Hydrogen Storage" Nanomaterials 13, no. 18: 2588. https://doi.org/10.3390/nano13182588