
First-Principles Investigation of Size Effects on Cohesive Energies of Transition-Metal Nanoclusters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
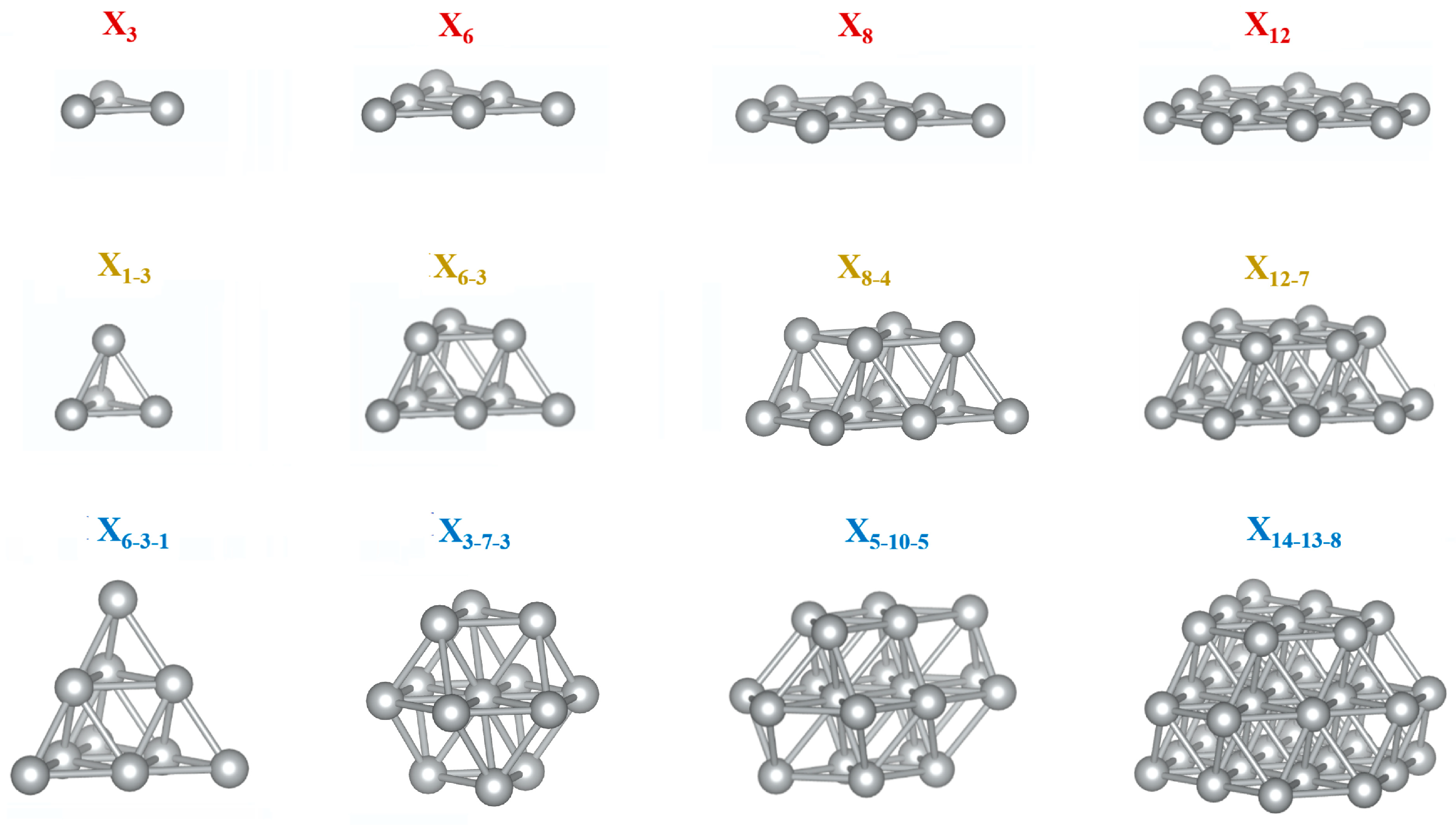
2. Computational Details
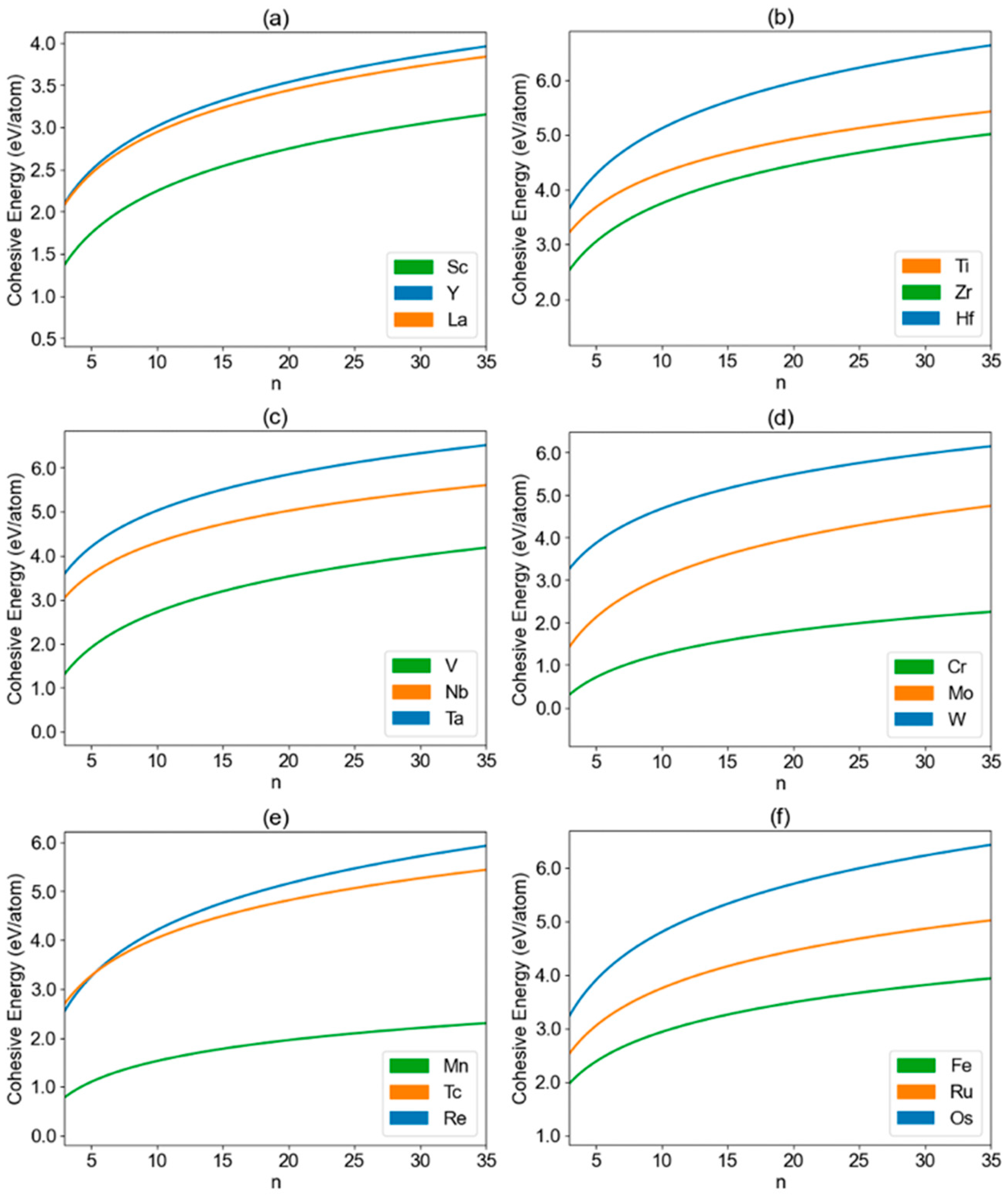
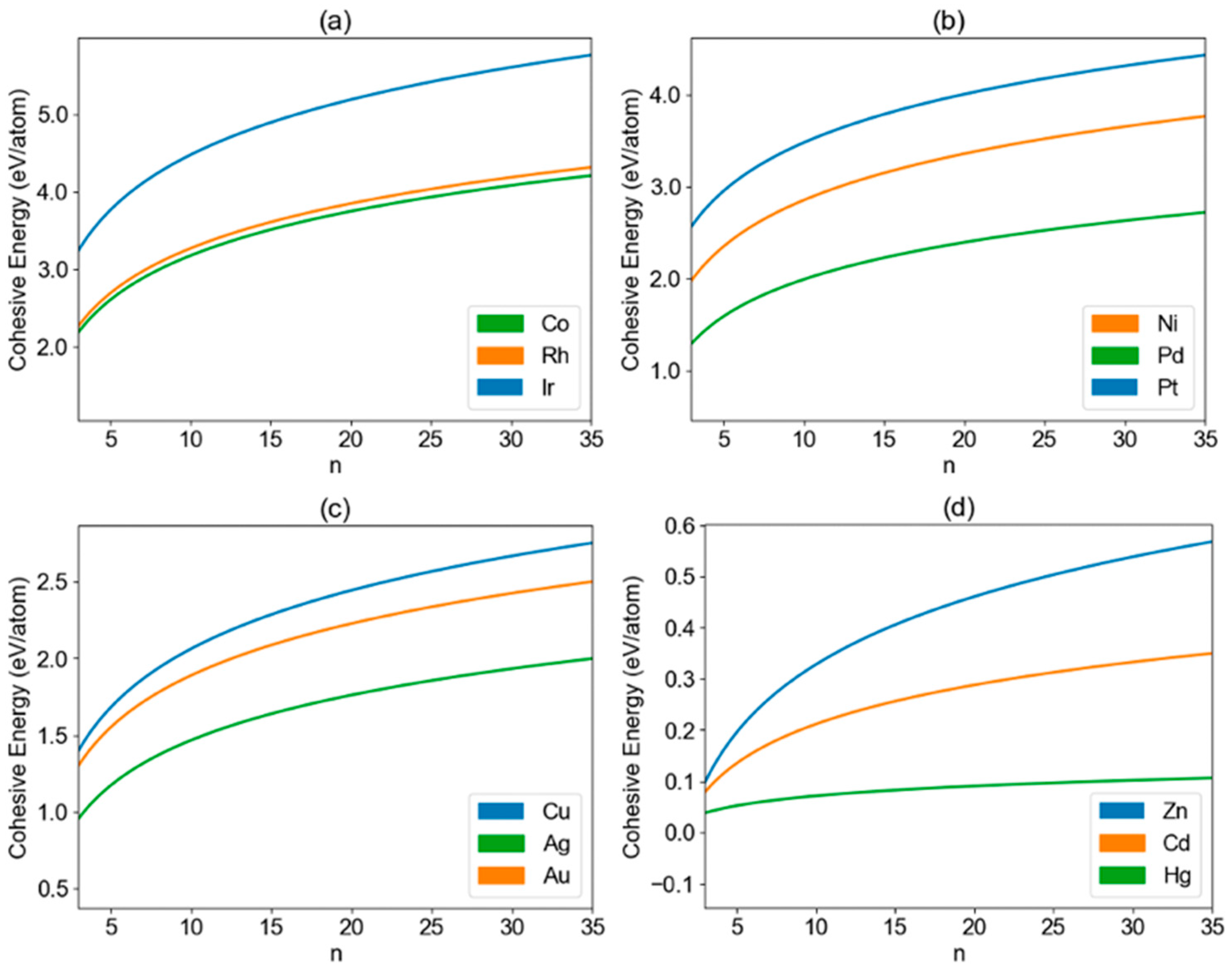
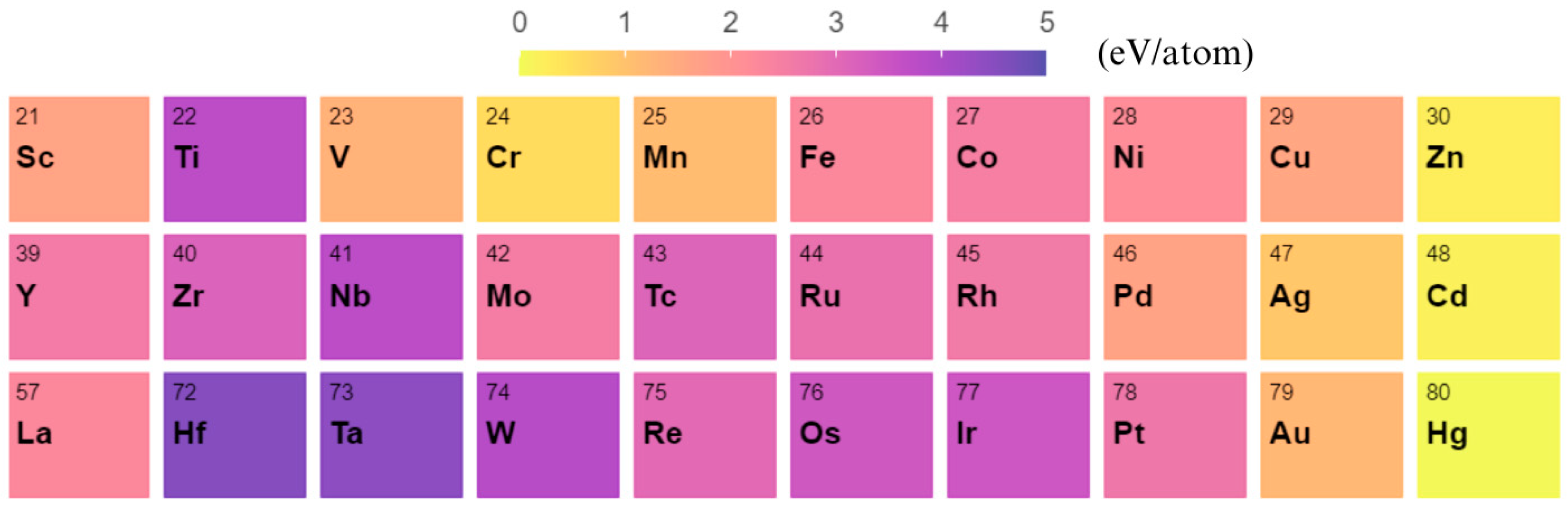
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neugebauer, N.; Wang, Y.; Elm, M.T.; Hofmann, D.M.; Heiliger, C.; Ye, X.; Klar, P.J. Distance and Size Dependence of the Interactions within Highly Ordered Magnetic Nanoparticle Mesocrystals. Phys. Rev. B 2023, 107, 184410. [Google Scholar] [CrossRef]
- Tyo, E.C.; Vajda, S. Catalysis by Clusters with Precise Numbers of Atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Meseguer, J.; Cabrero-Antonino, J.R.; Dominguez, I.; Leyva-Perez, A.; Corma, A. Small Gold Clusters Formed in Solution Give Reaction Turnover Numbers of 107 at Room Temperature. Science 2012, 338, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, W.; Xin, H.; Liu, R.; Wang, Q.; Cai, W.; Peng, X.; Yang, F.; Xin, H. Nanoparticles Advanced from Preclinical Studies to Clinical Trials for Lung Cancer Therapy. Cancer Nanotechnol. 2023, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.Y.; Zhang, S.P.; Liu, S.J. Significance and Implications of Nanoparticle-Biological Corona Fingerprints in Diagnosis, Prognosis, and Therapeutics for Diverse Disorders. Nanoscale 2023, 15, 11422–11433. [Google Scholar] [CrossRef]
- Thomas, T.; Kuttoth, H.; Nair, R.V.; Sandhyarani, N. Electrochemical Approach for the Synthesis of Ultrasmall Cu13 Clusters and Their Application in the Detection of Endotoxin. Langmuir 2023, 39, 10011–10020. [Google Scholar] [CrossRef]
- Rice, P.S.; Hu, P. Understanding Supported Noble Metal Catalysts Using First-Principles Calculations. J. Chem. Phys. 2019, 151, 180902. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, H.; Zhang, H.; Zhang, Y.; Shi, P.; Qu, K.; Cheng, S.B.; Wang, A.L.; Lu, Q. Filling Mesopores of Conductive Metal-Organic Frameworks with Cu Clusters for Selective Nitrate Reduction to Ammonia. ACS Appl. Mater. Interfaces 2022, 14, 32176–32182. [Google Scholar] [CrossRef]
- Oliveira, M.I.A.; Rivelino, R.; Mota, F.D.; Gueorguiev, G.K. Optical Properties and Quasiparticle Band Gaps of Transition-Metal Atoms Encapsulated by Silicon Cages. J. Phys. Chem. C 2014, 118, 5501–5509. [Google Scholar] [CrossRef]
- Gueorguiev, G.K.; Pacheco, J.M. Shapes of Cagelike Metal Carbide Clusters: First-Principles Calculations. Phys. Rev. B 2003, 68, 241401. [Google Scholar] [CrossRef]
- Liu, K.; Xing, F.L.; Xiao, Y.Y.; Yan, N.; Shimizu, K.; Furukawa, S. Development of a Highly Stable Ternary Alloy Catalyst for Dry Reforming of Methane. ACS Catal. 2023, 13, 3541–3548. [Google Scholar] [CrossRef]
- Jones, G.; Jakobsen, J.; Shim, S.; Kleis, J.; Andersson, M.; Rossmeisl, J.; Abildpedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B. First Principles Calculations and Experimental Insight into Methane Steam Reforming over Transition Metal Catalysts. J. Catal. 2008, 259, 147–160. [Google Scholar] [CrossRef]
- Wong, Y.J.; Halim, H.H.; Khairudin, N.F.; Pham, T.N.; Putra, S.E.M.; Hamamoto, Y.; Inagaki, K.; Hamada, I.; Mohamed, A.R.; Morikawa, Y. Dry Reforming of Methane on Cobalt Catalysts: DFT-Based Insights into Carbon Deposition versus Removal. J. Phys. Chem. C 2021, 125, 21902–21913. [Google Scholar] [CrossRef]
- Mizutani, U.; Inukai, M.; Sato, H.; Zijlstra, E.S. Physical Metallurgy; Laughlin, D.E., Hono, K., Eds.; Elsevier: Oxford, UK, 2014; pp. 103–202. [Google Scholar]
- Glasser, L.; Sheppard, D.A. Cohesive Energies and Enthalpies: Complexities, Confusions, and Corrections. Inorg. Chem. 2016, 55, 7103–7110. [Google Scholar] [CrossRef] [PubMed]
- Sachin; Pandey, B.K.; Jaiswal, R.L. Dimensional Effect on Cohesive Energy, Melting Temperature and Debye Temperature of Metallic Nanoparticles. Phys. B Condens. 2023, 651, 414602. [Google Scholar] [CrossRef]
- Qu, Y.D.; Liang, X.L.; Kong, X.Q.; Zhang, W.J. Size-Dependent Cohesive Energy, Melting Temperature, and Debye Temperature of Spherical Metallic Nanoparticles. Phys. Met. Metallogr. 2017, 118, 528–534. [Google Scholar] [CrossRef]
- Pabari, C. Size Dependent Properties of Metallic Nanoparticles. Mater. Today Proc. 2022, 55, 98–101. [Google Scholar] [CrossRef]
- Vanithakumari, S.C.; Nanda, K.K. A Universal Relation for the Cohesive Energy of Nanoparticles. Phys. Lett. A 2008, 372, 6930–6934. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Teeriniemi, J.; Melander, M.; Lipasti, S.; Hatz, R.; Laasonen, K. Fe-Ni Nanoparticles: A Multiscale First-Principles Study to Predict Geometry, Structure, and Catalytic Activity. J. Phys. Chem. C 2017, 121, 1667–1674. [Google Scholar] [CrossRef]
- Wang, S.J.; Kuang, X.Y.; Lu, C.; Li, Y.F.; Zhao, Y.R. Geometries, Stabilities, and Electronic Properties of Pt-Group-Doped Gold Clusters, Their Relationship to Cluster Size, and Comparison with Pure Gold Clusters. Phys. Chem. Chem. Phys. 2011, 13, 10119–10130. [Google Scholar] [CrossRef]
- Roy, G.; Chattopadhyay, A.P. Dissociation of Methane on Ni4 Cluster—A DFT Study. Comput. Theor. Chem. 2017, 1106, 7–14. [Google Scholar] [CrossRef]
- Cui, C.; Zhang, H.; Cheng, R.; Huang, B.; Luo, Z. On the Nature of Three-Atom Metal Cluster Catalysis for N2 Reduction to Ammonia. ACS Catal. 2022, 12, 14964–14975. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Curtarolo, S.; Setyawan, W.; Hart, G.L.W.; Jahnatek, M.; Chepulskii, R.V.; Taylor, R.H.; Wang, S.; Xue, J.; Yang, K.; Levy, O.; et al. AFLOW: An Automatic Framework for High-Throughput Materials Discovery. Comput. Mater. Sci. 2012, 58, 218–226. [Google Scholar] [CrossRef]
- Brunello, G.F.; Lee, J.H.; Lee, S.G.; Choi, J.I.; Harvey, D.; Jang, S.S. Interactions of Pt Nanoparticles with Molecular Components in Polymer Electrolyte Membrane Fuel Cells: Multi-Scale Modeling Approach. RSC Adv. 2016, 6, 69670–69676. [Google Scholar] [CrossRef]
- Lambie, S.; Steenbergen, K.G.; Gaston, N. Modulating the Thermal and Structural Stability of Gallenene via Variation of Atomistic Thickness. Nanoscale Adv. 2021, 3, 499–507. [Google Scholar] [CrossRef]
- Kim, H.K.; Huh, S.H.; Park, J.W.; Jeong, J.W.; Lee, G.H. The Cluster Size Dependence of Thermal Stabilities of Both Molybdenum and Tungsten Nanoclusters. Chem. Phys. Lett. 2002, 354, 165–172. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vig, A.; Doan, E.; Yang, K. First-Principles Investigation of Size Effects on Cohesive Energies of Transition-Metal Nanoclusters. Nanomaterials 2023, 13, 2356. https://doi.org/10.3390/nano13162356
Vig A, Doan E, Yang K. First-Principles Investigation of Size Effects on Cohesive Energies of Transition-Metal Nanoclusters. Nanomaterials. 2023; 13(16):2356. https://doi.org/10.3390/nano13162356
Chicago/Turabian StyleVig, Amogh, Ethan Doan, and Kesong Yang. 2023. "First-Principles Investigation of Size Effects on Cohesive Energies of Transition-Metal Nanoclusters" Nanomaterials 13, no. 16: 2356. https://doi.org/10.3390/nano13162356