
First-Principles Study of B16N16 Cluster-Assembled Porous Nanomaterials


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
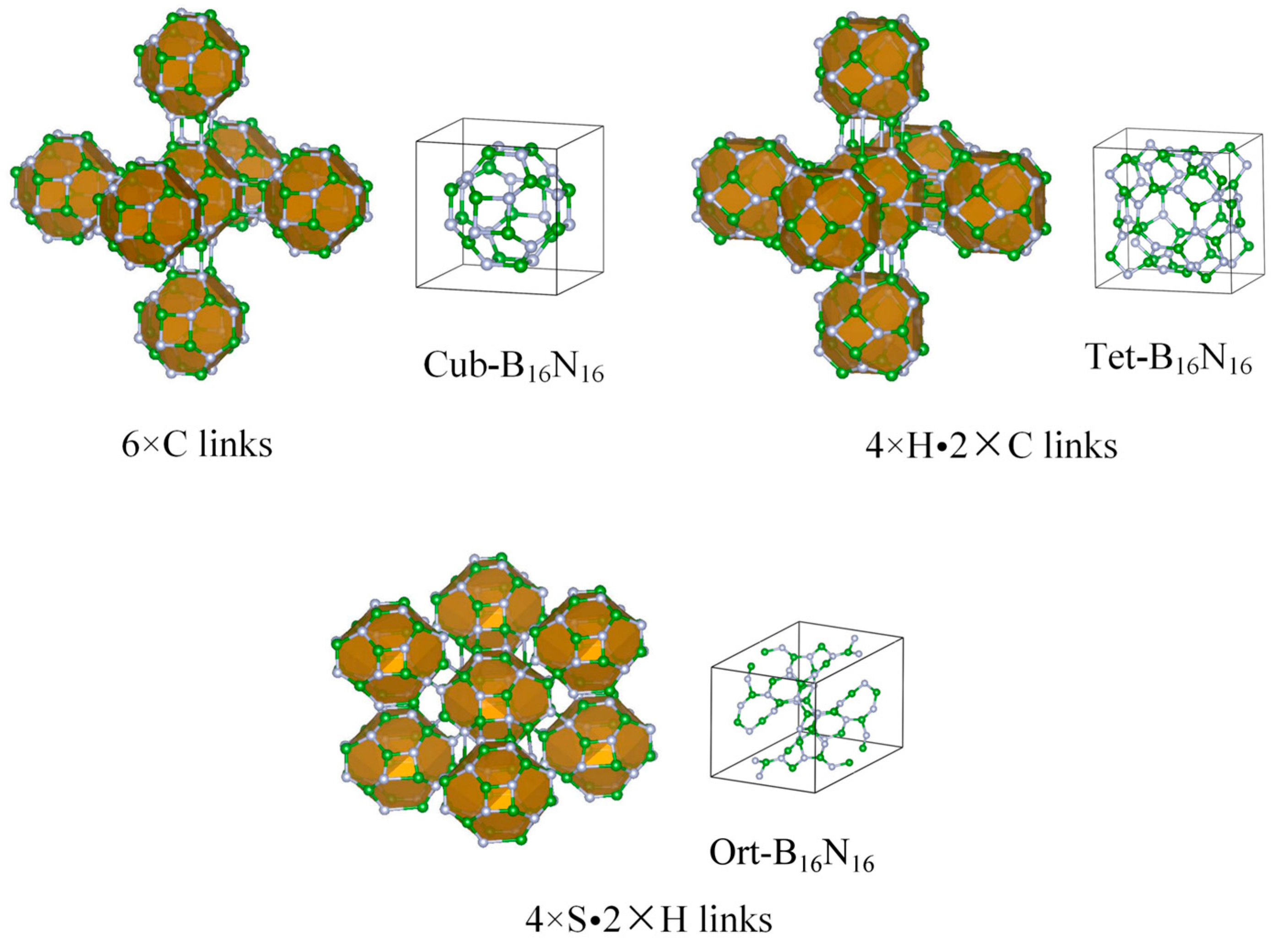
3.1. Structural Properties
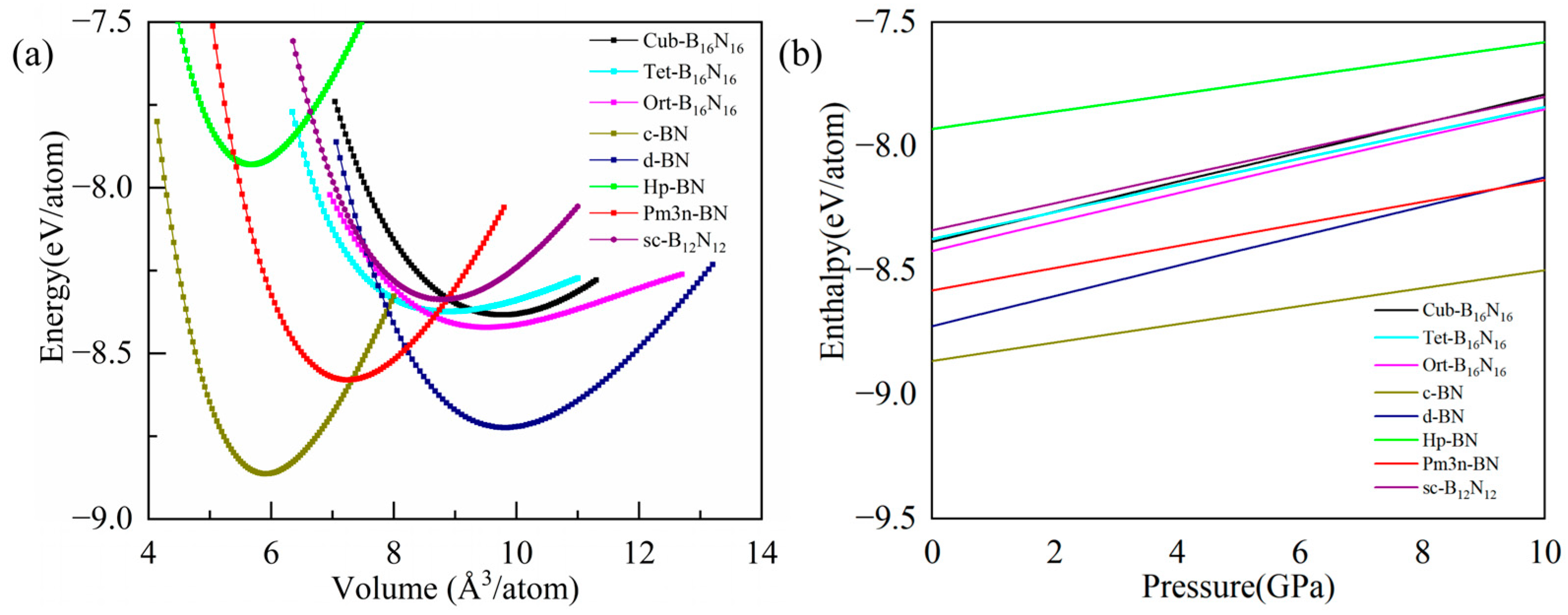
3.2. Stabilities
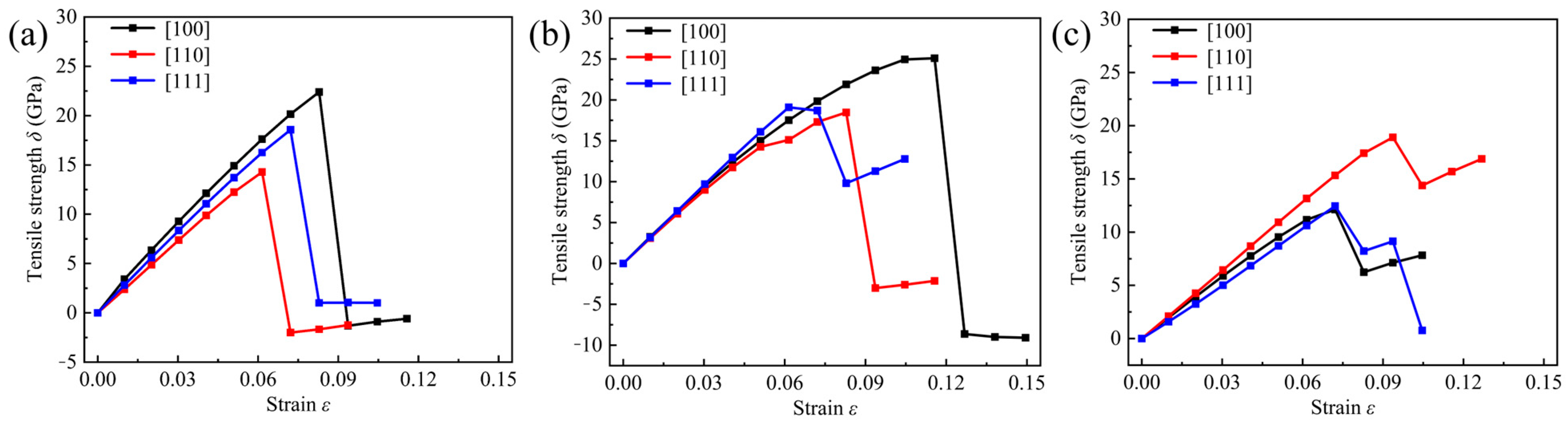
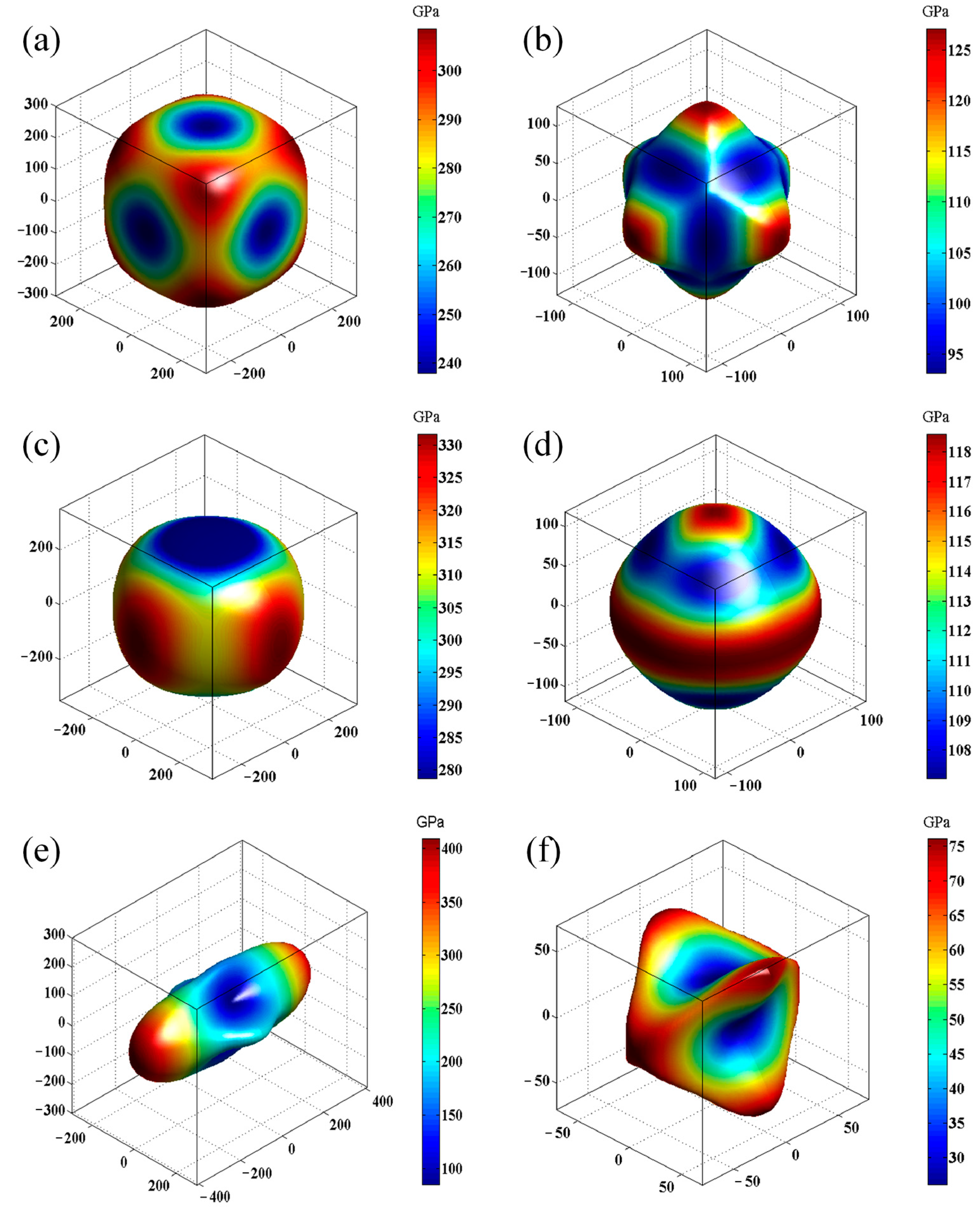
3.3. Mechanical Properties
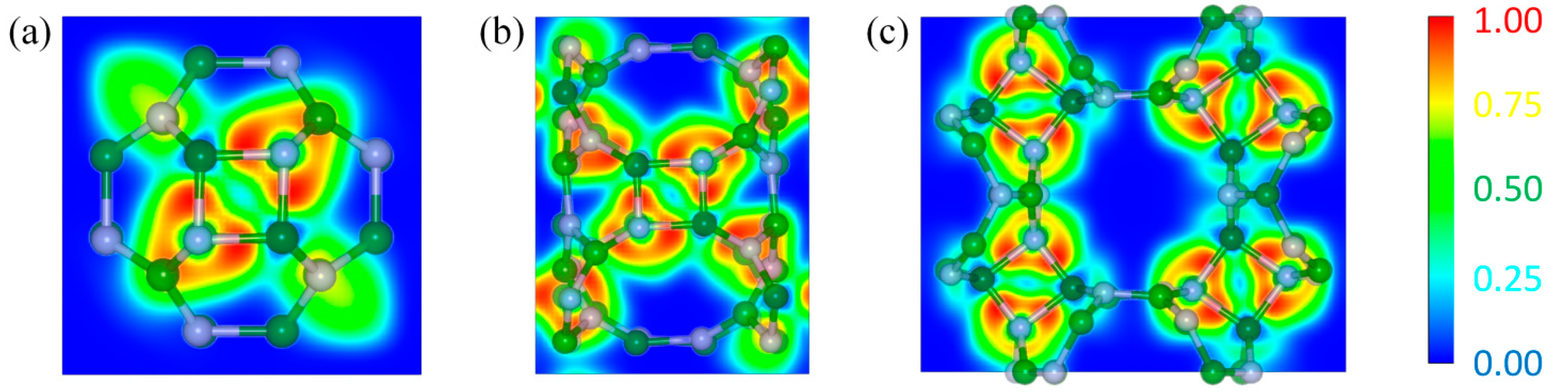
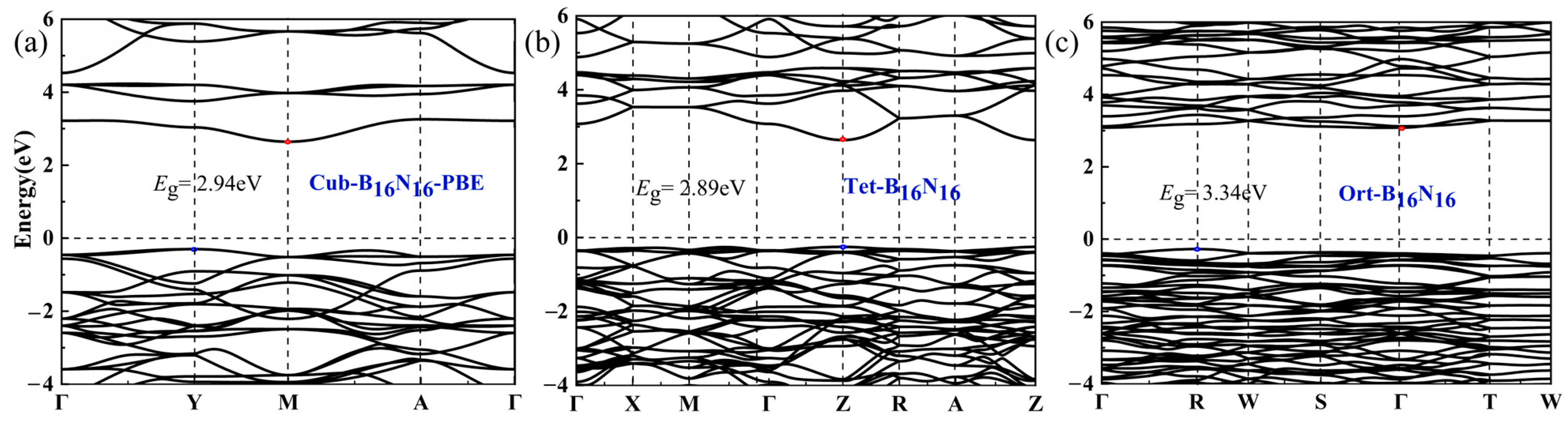
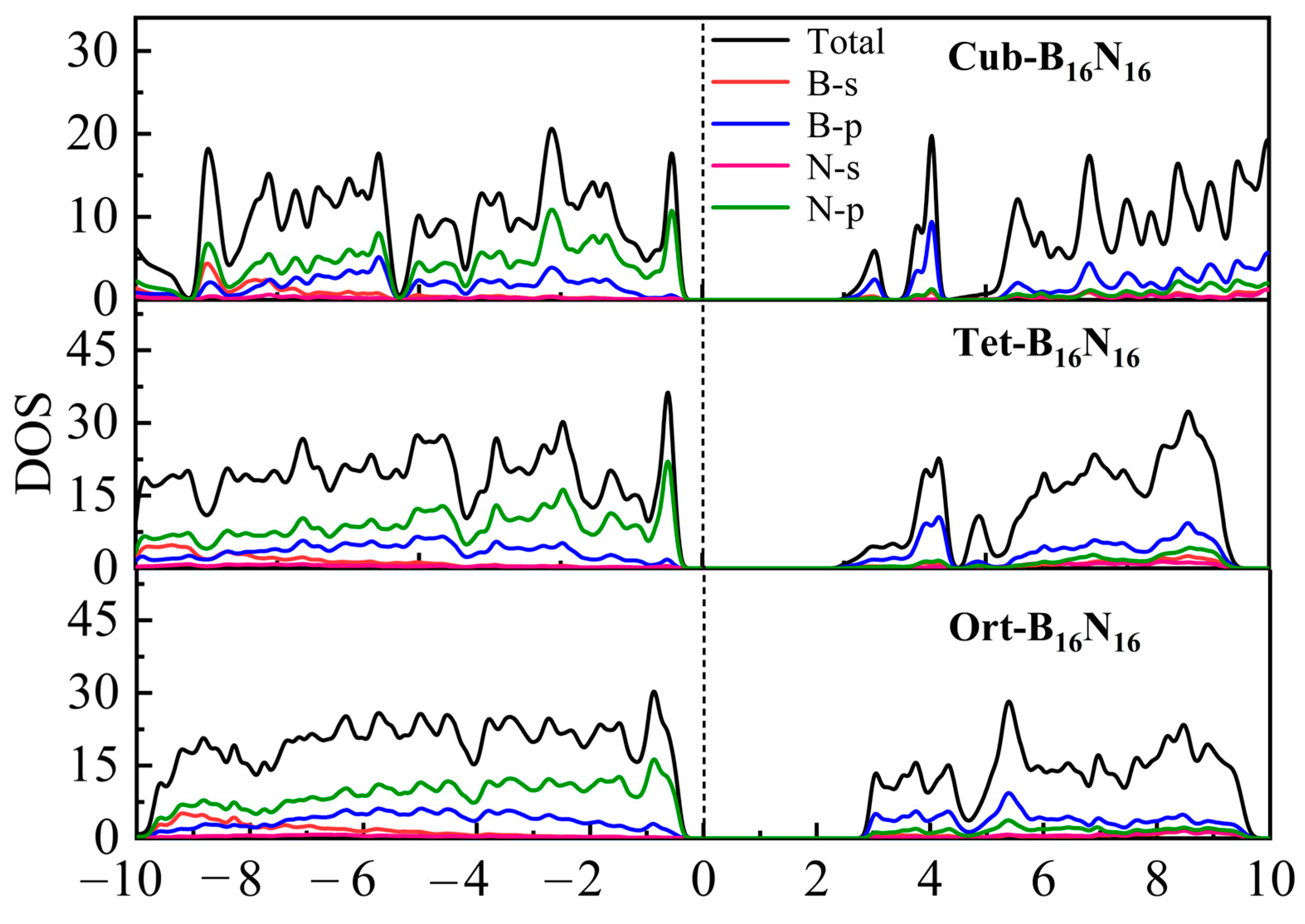
3.4. Electronic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berisha, A. Unraveling the electronic influence and nature of covalent bonding of aryl and alkyl radicals on the B12N12 nanocage cluster. Sci. Rep. 2023, 13, 752. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.J.; Yang, J.; Hou, J.G.; Zhu, Q. First-principles study of small-radius single-walled BN nanotubes. Phys. Rev. B 2003, 68, 035427. [Google Scholar] [CrossRef]
- Hao, J.; Li, L.; Gao, P.; Jiang, X.; Ban, C.; Shi, N. Boron nitride nanoribbons grown by chemical vapor deposition for VUV applications. Micromachines 2022, 13, 1372. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Cervenka, J.; Watanabe, K.; Taniguchi, T.; Chen, Y. Strong oxidation resistance of atomically thin boron nitride nanosheets. ACS Nano 2014, 8, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Z.; Song, T.; Cui, X. Sc-B24N24: A new low-density allotrope of BN. J. Phys. Chem. C 2022, 126, 12836–12844. [Google Scholar] [CrossRef]
- Oku, T.; Kuno, M.; Kitahara, H.; Narita, I. Formation, atomic structures and properties of boron nitride and carbon nanocage fullerene materials. Int. J. Inorg. Mater. 2001, 3, 597–612. [Google Scholar] [CrossRef]
- Oku, T.; Nishiwaki, A.; Narita, I.; Gonda, M. Formation and structure of B24N24 clusters. Chem. Phys. Lett. 2003, 380, 620–623. [Google Scholar] [CrossRef]
- Oku, T.; Nishiwaki, A.; Narita, I. Formation and atomic structure of B12N12 nanocage clusters studied by mass spectrometry and cluster calculation. Sci. Technol. Adv. Mater. 2004, 5, 635–638. [Google Scholar] [CrossRef]
- Oku, T.; Nishiwaki, A.; Narita, I. Formation and atomic structures of BnNn (n = 24–60) clusters studied by mass spectrometry, high-resolution electron microscopy and molecular orbital calculations. Phys. B 2004, 351, 184–190. [Google Scholar] [CrossRef]
- Stéphan, O.; Bando, Y.; Loiseau, A.; Willaime, F.; Shramchenko, N.; Tamiya, T.; Sato, T. Formation of small single-layer and nested BN cages under electron irradiation of nanotubes and bulk material. Appl. Phys. A 1998, 67, 107–111. [Google Scholar] [CrossRef]
- Seifert, G.; Fowler, P.W.; Mitchell, D.; Porezag, D.; Frauenheim, T. Boron-nitrogen analogues of the fullerenes: Electronic and structural properties. Chem. Phys. Lett. 1997, 268, 352. [Google Scholar] [CrossRef]
- Li, J.L.; He, T.; Yang, G.W. An all-purpose building block: B12N12 fullerene. Nanoscale 2012, 4, 1665–1670. [Google Scholar] [CrossRef]
- Yin, B.; Wang, G.; Sa, N.; Huang, Y. Bonding analysis and stability on alternant B16N16 cage and its dimers. J. Mol. Model. 2008, 14, 789–795. [Google Scholar] [CrossRef]
- Alexandre, S.S.; Chacham, H.; Nunes, R.W. Structure and energetics of boron nitride fullerenes: The role of stoichiometry. Phys. Rev. B 2001, 63, 045402. [Google Scholar] [CrossRef]
- Lan, Y.Z.; Cheng, W.D.; Wu, D.S.; Li, X.D.; Zhang, H.; Gong, Y.J.; Shen, J.; Li, F.F. Theoretical studies of third-order nonlinear optical response for B12N12, B24N24 and B36N36 clusters. J. Mol. Struc.-Theochem. 2005, 730, 9–15. [Google Scholar] [CrossRef]
- Wu, H.S.; Xu, X.H.; Strout, D.L.; Jiao, H. The structure and stability of B36N36 cages: A computational study. J. Mol. Model. 2006, 12, 1–8. [Google Scholar] [CrossRef]
- Strout, D.L. Fullerene-like cages versus alternant cages: Isomer stability of B13N13, B14N14, and B16N16. Chem. Phys. Lett. 2004, 383, 95–98. [Google Scholar] [CrossRef]
- Rogers, K.M.; Fowler, P.W.; Seifert, G. Chemical versus steric frustration in boron nitride heterofullerene polyhedral. Chem. Phys. Lett. 2000, 332, 43–50. [Google Scholar] [CrossRef]
- Xia, X.; Jelski, D.A.; Bowser, J.R.; George, T.F. MNDO Study of Boron-Nitrogen Analogues of Buckminsterfullerene. J. Am. Chem. Soc. 1992, 114, 6493–6496. [Google Scholar] [CrossRef]
- Khanna, S.N.; Jena, P. Assembling Crystals from Clusters. Phys. Rev. Lett. 1992, 69, 1664–1667. [Google Scholar] [CrossRef]
- Claridge, S.A.; Castleman, A.W., Jr.; Khanna, S.N.; Murray, C.B.; Sen, A.; Weiss, P.S. Cluster-Assembled Materials. ACS Nano 2009, 3, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Shi, J.; Zhou, A.; Cai, Y. A comparative investigation of sp3-hybridized Pm3n-BN and sc-B12N12 based on density functional theory (DFT). Mater. Today Commun. 2020, 25, 101582. [Google Scholar] [CrossRef]
- Alexandre, S.S.; Nunes, R.W.; Chacham, H. Energetics of the formation of dimers and solids of boron nitride fullerenes. Phys. Rev. B 2002, 66, 085406. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Cai, J.; Liu, G.; Zhou, P.; Wang, K.; Zhu, H. From the ZnO Hollow Cage Clusters to ZnO Nanoporous Phases: A First-Principles Bottom-Up Prediction. J. Phys. Chem. C 2013, 117, 17633–17643. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Effificient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Zhao, J.J.; Winey, J.M.; Gupta, Y.M. First-principles calculations of second- and third-order elastic constants for single crystals of arbitrary symmetry. Phys. Rev. B 2007, 75, 094105. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Brugger, K. Thermodynamic Definition of Higher Order Elastic Coefficients. Phys. Rev. 1964, 133, A1611–A1612. [Google Scholar] [CrossRef]
- Xu, S.; Wang, L.; Qiao, X.; Xu, X.; Cai, Y. Novel BN polymorphs transformed from the smallest BNNTs under high pressure. Comput. Mater. Sci. 2015, 110, 241–246. [Google Scholar] [CrossRef]
- Yu, J.; Qu, L.; Veen, E.; Katsnelson, M.I.; Yuan, S. Hyperhoneycomb boron nitride with anisotropic mechanical, electronic, and optical properties. Phys. Rev. Mater. 2017, 1, 045001. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zeng, L.; Zhang, Y.; Xu, X. Multiporous sp2-hybridized boron nitride (d-BN): Stability, mechanical properties, lattice thermal conductivity and promising application in energy storage. Phys. Chem. Chem. Phys. 2018, 20, 20726–20731. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. In. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Ohba, N.; Miwa, K.; Nagasako, N.; Fukumoto, A. First-principles study on structural, dielectric, and dynamical properties for three BN polytypes. Phys. Rev. B 2001, 63, 115207. [Google Scholar] [CrossRef]
- Peng, Q.; Ji, W.; De, S. Mechanical properties of the hexagonal boron nitride monolayer: Ab initio study. Comp. Mater. Sci. 2012, 56, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Nag, A.; Raidongia, K.; Hembram, K.P.S.S.; Datta, R.; Waghmare, U.V.; Rao, C.N.R. Graphene analogues of BN: Novel synthesis and properties. ACS Nano 2010, 4, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Soma, T.; Sawaoka, S.; Saito, S. Characterization of wurtzite type boron nitride synthesized by shock compression. Mater. Res. Bull. 1974, 9, 755–762. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- He, C.; Sun, L.; Zhang, C.; Peng, X.; Zhang, K.; Zhong, J. Z-BN: A novel superhard boron nitride phase. Phys. Chem. Chem. Phys. 2012, 14, 10967–10971. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Šimůnek, A.; Vackář, J. Hardness of covalent and ionic crystals: First-principle calculations. Phys. Rev. Lett. 2006, 96, 085501. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, J.; Illas, F.; Bromley, S.T. Ultralow-density nanocage-based metal-oxide polymorphs. Phys. Rev. Lett. 2007, 99, 235502. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | SG | a(Å) | b(Å) | c(Å) | V | ρ | Etot(eV) | Eg(eV) | Eg/Eg,c | |
|---|---|---|---|---|---|---|---|---|---|---|
| Cub-B16N16 | 6.772 | 6.772 | 6.772 | 9.77 | 2.124 | −8.38 | 2.94 | 0.659 | ||
| Tet-B16N16 | P4/nbm | 8.991 | 8.991 | 6.857 | 8.89 | 2.379 | −8.37 | 2.80 | 0.628 | |
| Ort-B16N16 | Imma | 8.975 | 12.669 | 10.726 | 9.51 | 2.163 | −8.42 | 3.34 | 0.749 | |
| c-BN | This work | 3.615 | 3.615 | 3.615 | 5.90 | 3.489 | −8.86 | 4.46 | 1.00 | |
| Cal. [22] | 3.625 | 3.625 | 3.625 | −9.37 | ||||||
| Expt. [38] | 3.615 | 3.615 | 3.615 | 3.489 | 6.1~6.4 | |||||
| h-BN(2D) | This work | P63/mmc | 2.506 | 2.506 | −8.16 | 3.95 | 0.89 | |||
| Cal. [39] | 2.512 | 2.512 | ||||||||
| Expt. [40] | 2.490 | 2.490 | ||||||||
| w-BN | This work | P63mc | 2.549 | 2.549 | 4.231 | 5.92 | 2.095 | −8.85 | 5.20 | 1.17 |
| Cal. [22] | 2.555 | 2.555 | 4.225 | −9.35 | ||||||
| Expt. [41] | 2.553 | 2.553 | 4.228 | |||||||
| d-BN | This work | 12.290 | 12.290 | 12.290 | 9.81 | 2.101 | −8.72 | 4.84 | 1.09 | |
| Cal. [36] | 12.292 | 12.292 | 12.292 | 2.130 | 4.86 | |||||
| Hp-BN | This work | P6222 | 2.600 | 2.600 | 5.811 | 5.67 | 3.633 | −7.93 | 3.70 | 0.83 |
| Cal. [35] | 2.610 | 2.610 | 5.828 | −7.78 | 3.45 | |||||
| Pm3n-BN | This work | 4.428 | 4.428 | 4.428 | 7.23 | 2.849 | −8.58 | 4.55 | 1.02 | |
| Cal. [22] | 4.418 | 4.418 | 4.418 | 2.868 | −8.33 | 4.53 | ||||
| sc-B12N12 | This work | 11.819 | 11.819 | 11.819 | 8.75 | 2.345 | −8.33 | 4.98 | 1.12 | |
| Cal. [22] | 11.819 | 11.819 | 11.819 | 2.396 | −8.20 | 5.02 |
| Structure | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | K | Y | Hv |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cub-B16N16 | 304 | 118 | 127 | 180 | 112 | 1.61 | 279 | 15.19 | ||||||
| Tet-B16N16 | 376 | 84 | 122 | 314 | 123 | 134 | 197 | 123 | 1.60 | 305 | 16.30 | |||
| Ort-B16N16 | 225 | 73 | 54 | 480 | 112 | 200 | 76 | 26 | 76 | 141 | 69 | 2.07 | 178 | 8.17 |
| c-BN | 797 | 175 | 456 | 382 | 391 | 0.98 | 875 | 63.37 | ||||||
| Cal. [44] | 780 | 173 | 444 | 376 | 382 | 62.82 | ||||||||
| d-BN | 300 | 175 | 120 | 216 | 93 | 2.32 | 243 | 7.38 | ||||||
| Cal. [36] | 252 | 111 | 2.17 | |||||||||||
| Hp-BN | 873 | 154 | 360 | 384 | 366 | 1.04 | 832 | 55.97 | ||||||
| Cal. [35] | 892 | 166 | 363 | 375 | ||||||||||
| Pm3n-BN | 712 | 90 | 195 | 297 | 235 | 1.26 | 558 | 33.74 | ||||||
| Cal. [22] | 781 | 116 | 218 | 337 | 218~332 | |||||||||
| sc-B12N12 | 452 | 127 | 163 | 232 | 162 | 0.69 | 391 | 17.28 | ||||||
| Cal. [22] | 483 | 160 | 190 | 268 | 162~190 |
| Structure | Direction | Ideal Tensile Strength (GPa) | Maximum Strain (%) |
|---|---|---|---|
| Cub-B16N16 | [100] | 22.39 | 8 |
| [110] | 14.29 | 6 | |
| [111] | 18.56 | 7 | |
| Tet-B16N16 | [100] | 25.09 | 12 |
| [110] | 18.47 | 8 | |
| [111] | 18.69 | 7 | |
| Ort-B16N16 | [100] | 12.12 | 7 |
| [110] | 18.91 | 9 | |
| [111] | 12.45 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, X.; Liu, L.; Song, T.; Liu, Z.; Cui, X. First-Principles Study of B16N16 Cluster-Assembled Porous Nanomaterials. Nanomaterials 2023, 13, 1927. https://doi.org/10.3390/nano13131927
Wang X, Zhang X, Liu L, Song T, Liu Z, Cui X. First-Principles Study of B16N16 Cluster-Assembled Porous Nanomaterials. Nanomaterials. 2023; 13(13):1927. https://doi.org/10.3390/nano13131927
Chicago/Turabian StyleWang, Xin, Xiaoyue Zhang, Liwei Liu, Tielei Song, Zhifeng Liu, and Xin Cui. 2023. "First-Principles Study of B16N16 Cluster-Assembled Porous Nanomaterials" Nanomaterials 13, no. 13: 1927. https://doi.org/10.3390/nano13131927