
Electronic Structures of Monolayer Binary and Ternary 2D Materials: MoS2, WS2, Mo1−xCrxS2, and W1−xCrxS2 Using Density Functional Theory Calculations

Abstract
:1. Introduction
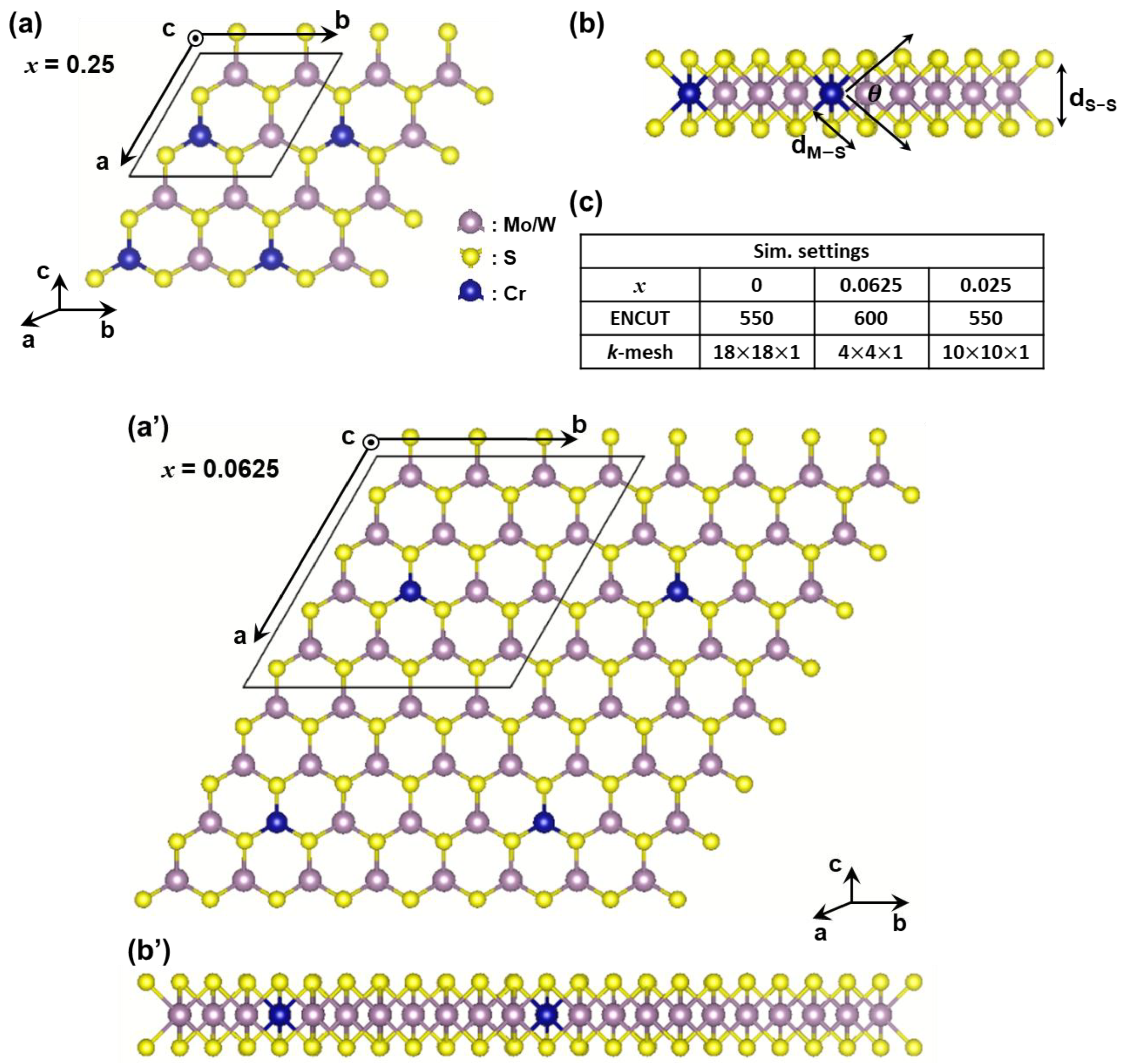
2. Materials and Methods
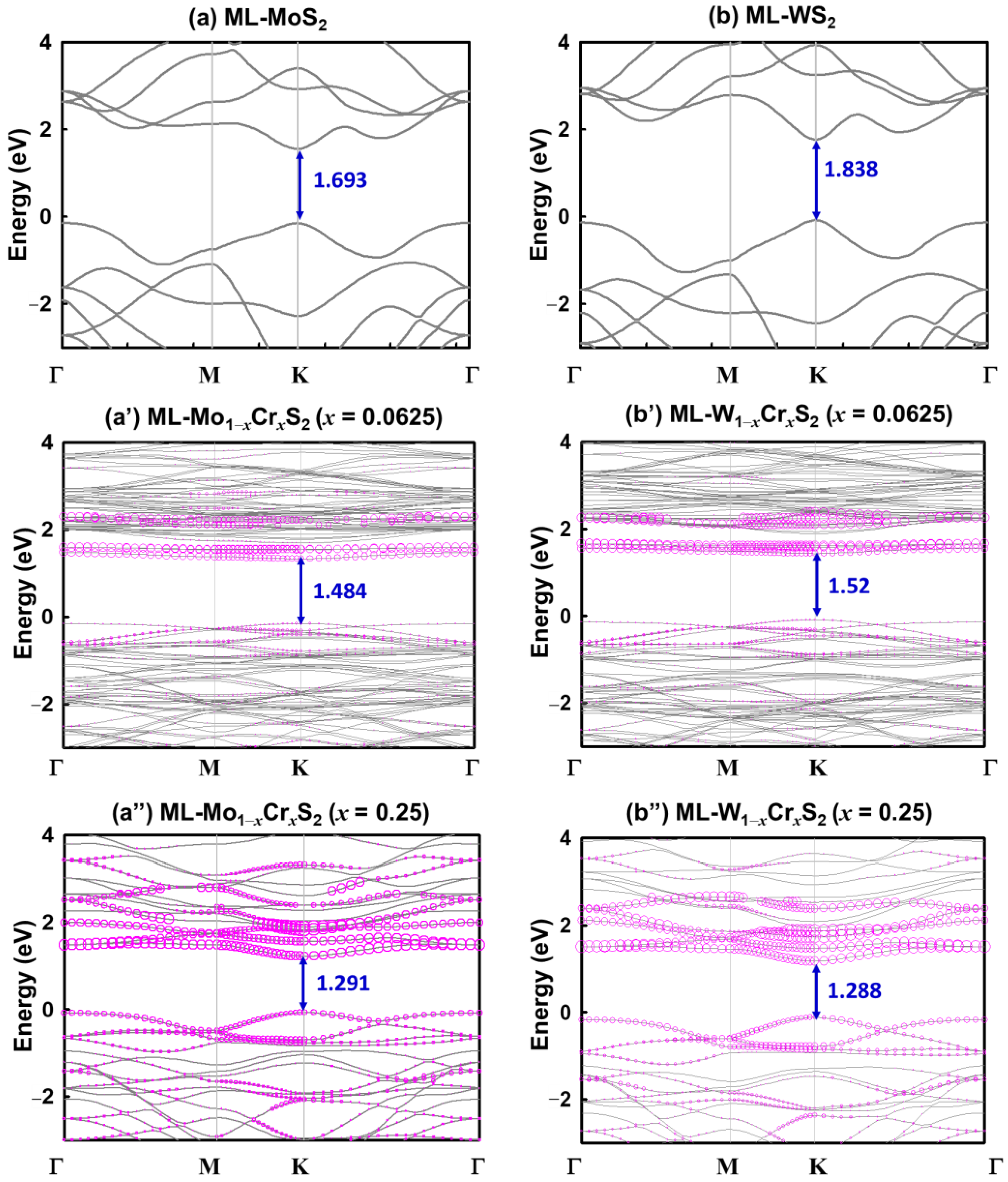
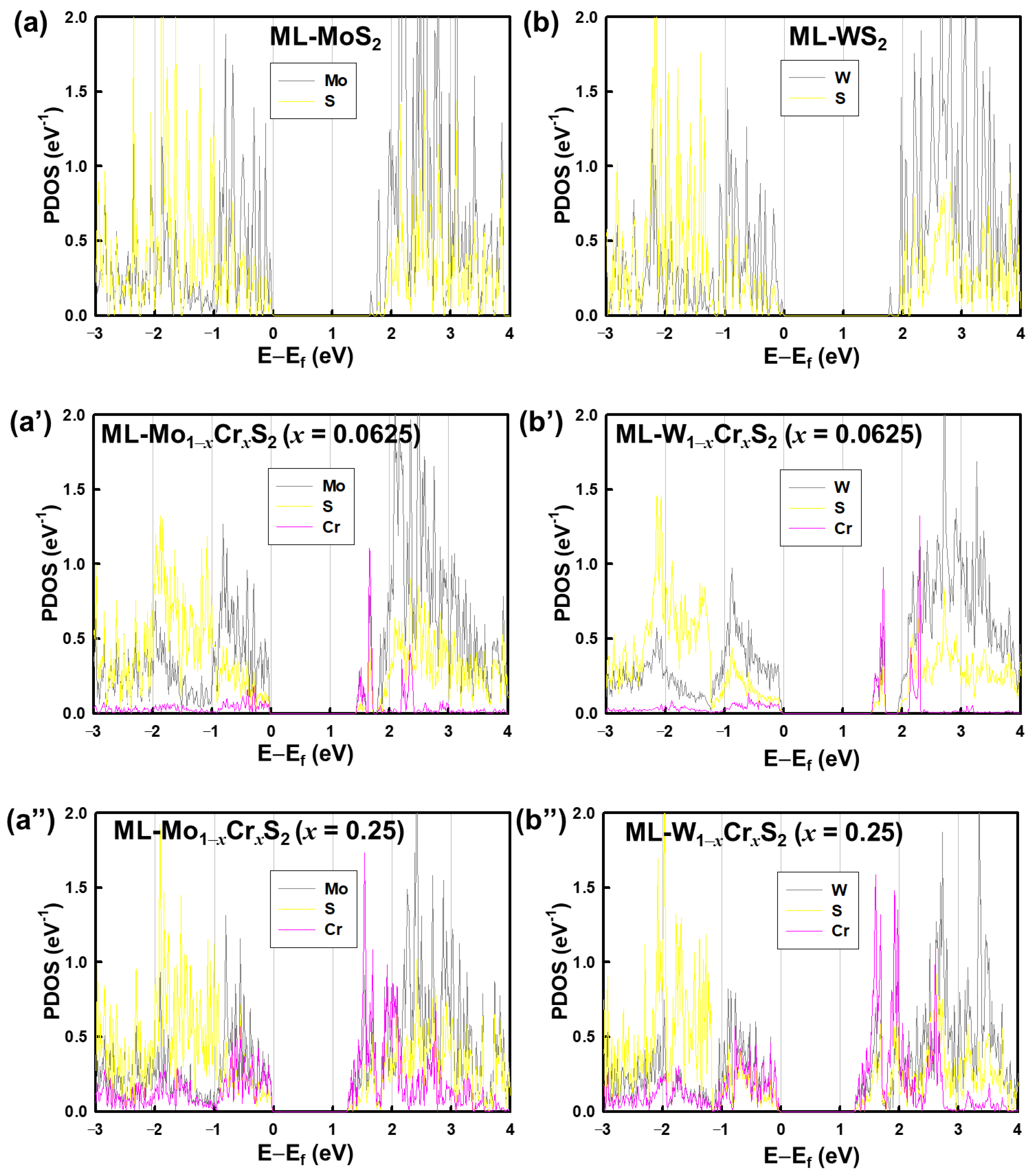
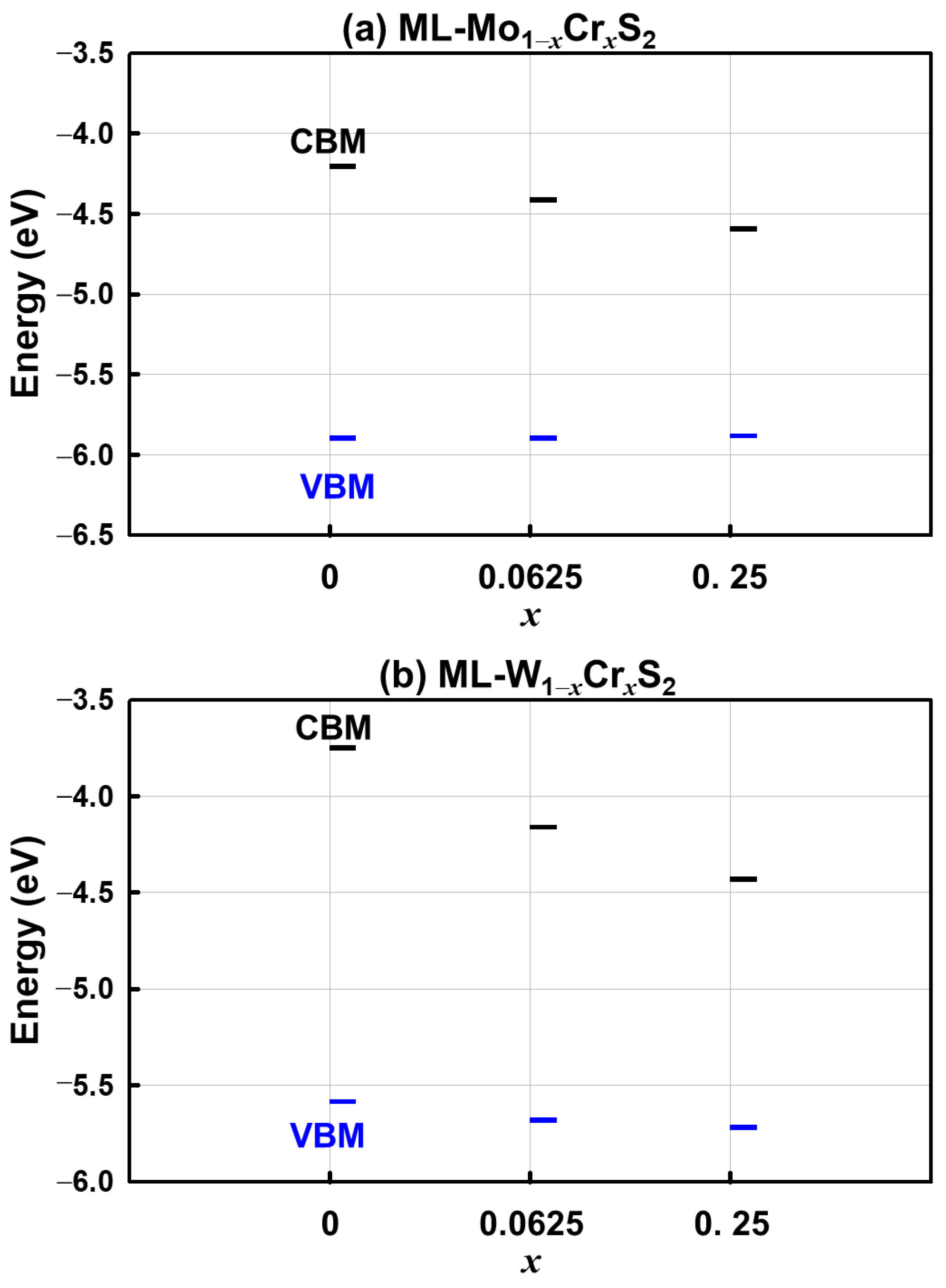
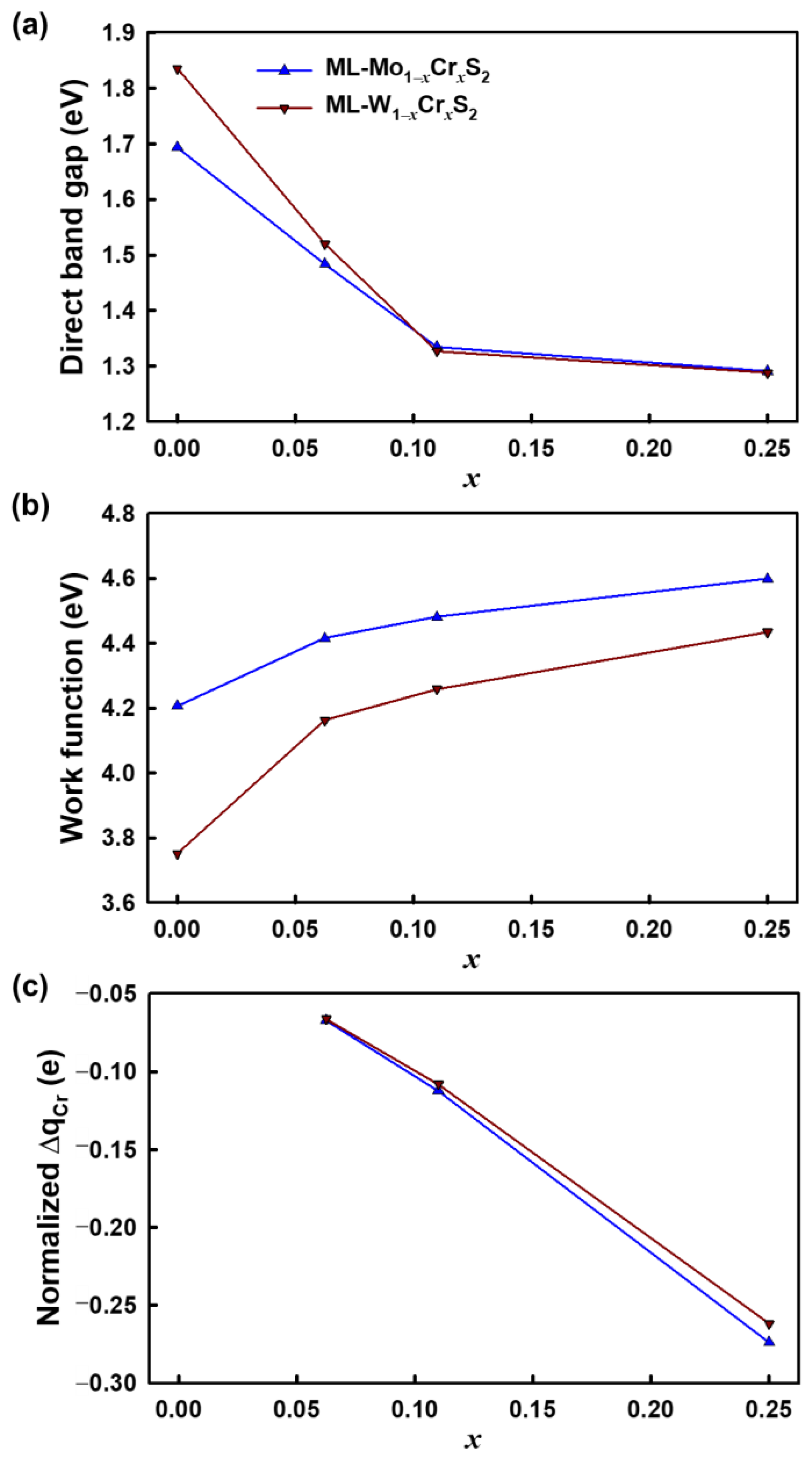
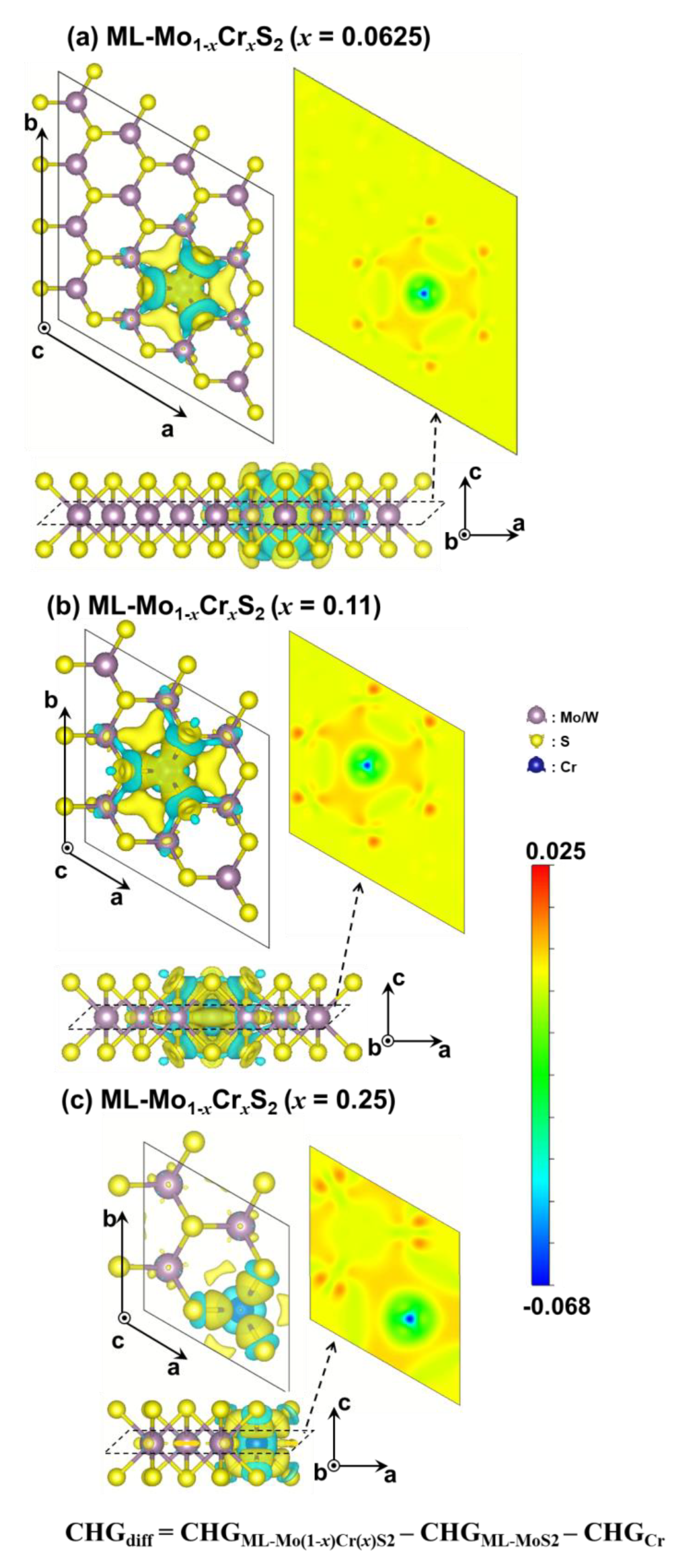
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zeng, X.; Xiao, C.; Liao, L.; Tu, Z.; Lai, Z.; Xiong, K.; Wen, Y. Two-Dimensional (2D) TM-Tetrahydroxyquinone Metal–Organic Framework for Selective CO2 Electrocatalysis: A DFT Investigation. Nanomaterials 2022, 12, 4049. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Boulet, P.; Record, M.-C. Structure–Property Relationships in Transition Metal Dichalcogenide Bilayers under Biaxial Strains. Nanomaterials 2021, 11, 2639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Record, M.-C.; Boulet, P. Electron Density and Its Relation with Electronic and Optical Properties in 2D Mo/W Dichalcogenides. Nanomaterials 2020, 10, 2221. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Ubrig, N.; Berger, H.; Kuzmenko, A.B.; Morpurgo, A.F. Mono- and Bilayer WS2 Light-Emitting Transistors. Nano Lett. 2014, 14, 2019–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.W.; Iqbal, M.Z.; Khan, M.F.; Shehzad, M.A.; Seo, Y.; Park, J.H.; Hwang, C.; Eom, J. High-Mobility and Air-Stable Single-Layer WS2 Field-Effect Transistors Sandwiched between Chemical Vapor Deposition-Grown Hexagonal BN Films. Sci. Rep. 2015, 5, 10699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radisavljevic, B.; Whitwick, M.B.; Kis, A. Integrated Circuits and Logic Operations Based on Single-Layer MoS2. ACS Nano 2011, 5, 9934–9938. [Google Scholar] [CrossRef]
- Pu, J.; Yomogida, Y.; Liu, K.-K.; Li, L.-J.; Iwasa, Y.; Takenobu, T. Highly Flexible MoS2 Thin-Film Transistors with Ion Gel Dielectrics. Nano Lett. 2012, 12, 4013–4017. [Google Scholar] [CrossRef]
- Island, J.O.; Kuc, A.; Diependaal, E.H.; Bratschitsch, R.; Zant, H.S.J.; van der Heine, T.; Castellanos-Gomez, A. Precise and Reversible Band Gap Tuning in Single-Layer MoSe2 by Uniaxial Strain. Nanoscale 2016, 8, 2589–2593. [Google Scholar] [CrossRef] [Green Version]
- Gusakova, J.; Wang, X.; Shiau, L.L.; Krivosheeva, A.; Shaposhnikov, V.; Borisenko, V.; Gusakov, V.; Tay, B.K. Electronic Properties of Bulk and Monolayer TMDs: Theoretical Study Within DFT Framework (GVJ-2e Method). Phys. Status Solidi 2017, 214, 1700218. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef]
- Tongay, S.; Fan, W.; Kang, J.; Park, J.; Koldemir, U.; Suh, J.; Narang, D.S.; Liu, K.; Ji, J.; Li, J.; et al. Tuning Interlayer Coupling in Large-Area Heterostructures with CVD-Grown MoS2 and WS2 Monolayers. Nano Lett. 2014, 14, 3185–3190. [Google Scholar] [CrossRef]
- Siao, M.-D.; Lin, Y.-C.; He, T.; Tsai, M.-Y.; Lee, K.-Y.; Chang, S.-Y.; Lin, K.-I.; Lin, Y.-F.; Chou, M.-Y.; Suenaga, K.; et al. Embedment of Multiple Transition Metal Impurities into WS2 Monolayer for Bandstructure Modulation. Small 2021, 17, e2007171. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, X.; Gu, Z.; Ling, T.; Li, Y.; Ma, S. Cu-Doped MoSe2 Monolayer: A Novel Candidate for Dissolved Gas Analysis in Transformer Oil. ACS Omega 2020, 5, 30603–30609. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, Z. Co-Modulation of Graphene by N Hetero-Doping &vacancy Defects and the Effect on NO2 Adsorption and Sensing: First-Principles Study. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 116, 113737. [Google Scholar] [CrossRef]
- Shakil, M.; Naz, A.; Zeba, I.; Gillani, S.S.A.; Rafique, M.; Ahmed, S.; Zafar, M. Structural and Magnetic Behavior of MoS2 on Doping of Transition Metals: A DFT Study. J. Supercond. Nov. Magn. 2021, 34, 3441–3453. [Google Scholar] [CrossRef]
- Andriotis, A.N.; Menon, M. Tunable Magnetic Properties of Transition Metal Doped MoS2. Phys. Rev. B 2014, 90, 125304. [Google Scholar] [CrossRef] [Green Version]
- Mohana Roopan, S.; Khan, M.A. MoS2 Based Ternary Composites: Review on Heterogeneous Materials as Catalyst for Photocatalytic Degradation. Catal. Rev. 2021, 1–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, H.; Du, S.; Yang, X. Conversion of MoS2 to a Ternary MoS2–xSex Alloy for High-Performance Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 11327–11337. [Google Scholar] [CrossRef]
- Wang, L.; Hu, P.; Long, Y.; Liu, Z.; He, X. Recent Advances in Ternary Two-Dimensional Materials: Synthesis, Properties and Applications. J. Mater. Chem. A 2017, 5, 22855–22876. [Google Scholar] [CrossRef]
- Gao, W.; Zheng, Z.; Wen, P.; Huo, N.; Li, J. Novel Two-Dimensional Monoelemental and Ternary Materials: Growth, Physics and Application. Nanophotonics 2020, 9, 2147–2168. [Google Scholar] [CrossRef]
- Gao, T.; Zhang, Q.; Li, L.; Zhou, X.; Li, L.; Li, H.; Zhai, T. 2D Ternary Chalcogenides. Adv. Opt. Mater. 2018, 6, 1800058. [Google Scholar] [CrossRef]
- Xi, J.; Zhao, T.; Wang, D.; Shuai, Z. Tunable Electronic Properties of Two-Dimensional Transition Metal Dichalcogenide Alloys: A First-Principles Prediction. J. Phys. Chem. Lett. 2014, 5, 285–291. [Google Scholar] [CrossRef]
- Chen, Y.; Xi, J.; Dumcenco, D.O.; Liu, Z.; Suenaga, K.; Wang, D.; Shuai, Z.; Huang, Y.-S.; Xie, L. Tunable Band Gap Photoluminescence from Atomically Thin Transition-Metal Dichalcogenide Alloys. ACS Nano 2013, 7, 4610–4616. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Li, Y. Numerical Calculation of Electronic Properties of Transition Metal-Doped MWS2 via DFT. In Proceedings of the Scientific Computing in Electrical Engineering, Eindhoven, The Netherlands, 16–20 February 2020; van Beurden, M., Budko, N., Schilders, W., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 53–61. [Google Scholar]
- Tsai, Y.-C.; Li, Y. On Electronic Structure and Geometry of MoX2 (X = S, Se, Te) and Black Phosphorus by Ab Initio Simulation with Various van Der Waals Corrections. In Proceedings of the 2017 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD), Kamakura, Japan, 7–9 September 2017; pp. 169–172. [Google Scholar]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B Condens. Matter. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Thajitr, W.; Busayaporn, W.; Rai, D.P.; Sukkabot, W. Modulation of Electronic and Magnetic Properties of MoX2 (X = S and Se) Monolayer via Mono- and Co-Transition Metal Dopants: Spin Density Functional Theory. Phys. Scr. 2022, 97, 095805. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Explored Material | ML-Mo1−xCrxS2 | ML-W1−xCrxS2 | ||||
|---|---|---|---|---|---|---|
| Mole Fraction x | 0 | 0.0625 | 0.25 | 0 | 0.0625 | 0.25 |
| a (Å) | 3.18 | 12.697 | 6.299 | 3.181 | 12.705 | 6.307 |
| Normalized a (Å) | 3.18 | 3.174 | 3.149 | 3.181 | 3.176 | 3.154 |
| dM−S (Å) | 2.413 | 2.325 | 2.329 | 2.419 | 2.334 | 2.338 |
| Thickness (Å) | 3.131 | 3.143 | 3.169 | 3.148 | 3.152 | 3.169 |
| (deg) | 80.9 | 81.683 | 81.983 | 81.185 | 81.7 | 82.086 |
| Eform (eV) | 0.19 | 0.75 | 0.31 | 1.22 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Li, Y.; Chuang, M.-H. Electronic Structures of Monolayer Binary and Ternary 2D Materials: MoS2, WS2, Mo1−xCrxS2, and W1−xCrxS2 Using Density Functional Theory Calculations. Nanomaterials 2023, 13, 68. https://doi.org/10.3390/nano13010068
Chen C-Y, Li Y, Chuang M-H. Electronic Structures of Monolayer Binary and Ternary 2D Materials: MoS2, WS2, Mo1−xCrxS2, and W1−xCrxS2 Using Density Functional Theory Calculations. Nanomaterials. 2023; 13(1):68. https://doi.org/10.3390/nano13010068
Chicago/Turabian StyleChen, Chieh-Yang, Yiming Li, and Min-Hui Chuang. 2023. "Electronic Structures of Monolayer Binary and Ternary 2D Materials: MoS2, WS2, Mo1−xCrxS2, and W1−xCrxS2 Using Density Functional Theory Calculations" Nanomaterials 13, no. 1: 68. https://doi.org/10.3390/nano13010068