
X-ray Diffraction Imaging of Deformations in Thin Films and Nano-Objects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
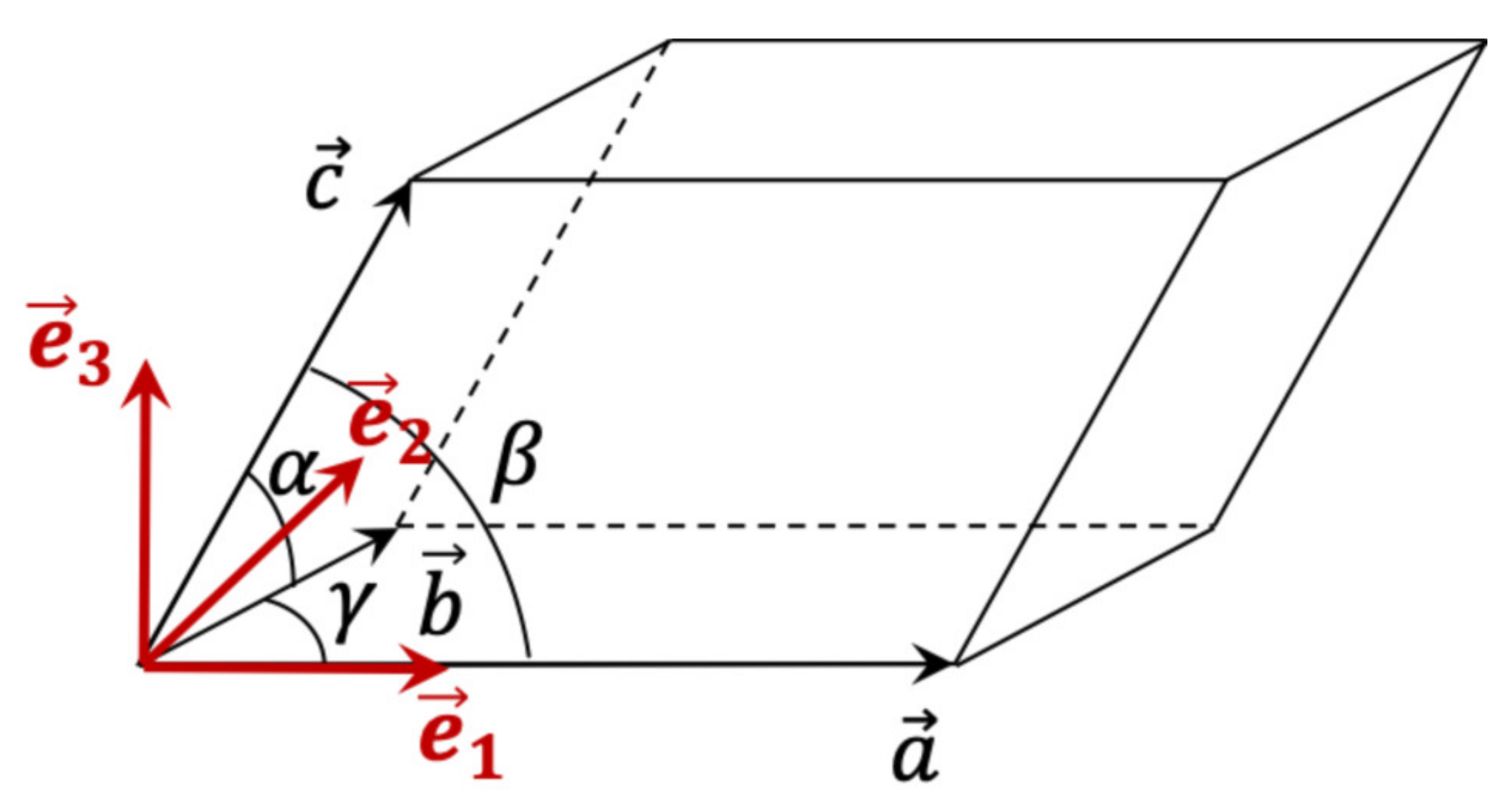
2. X-ray Diffraction by a Distorted Crystal
2.1. Kinematic Theory—Reminder
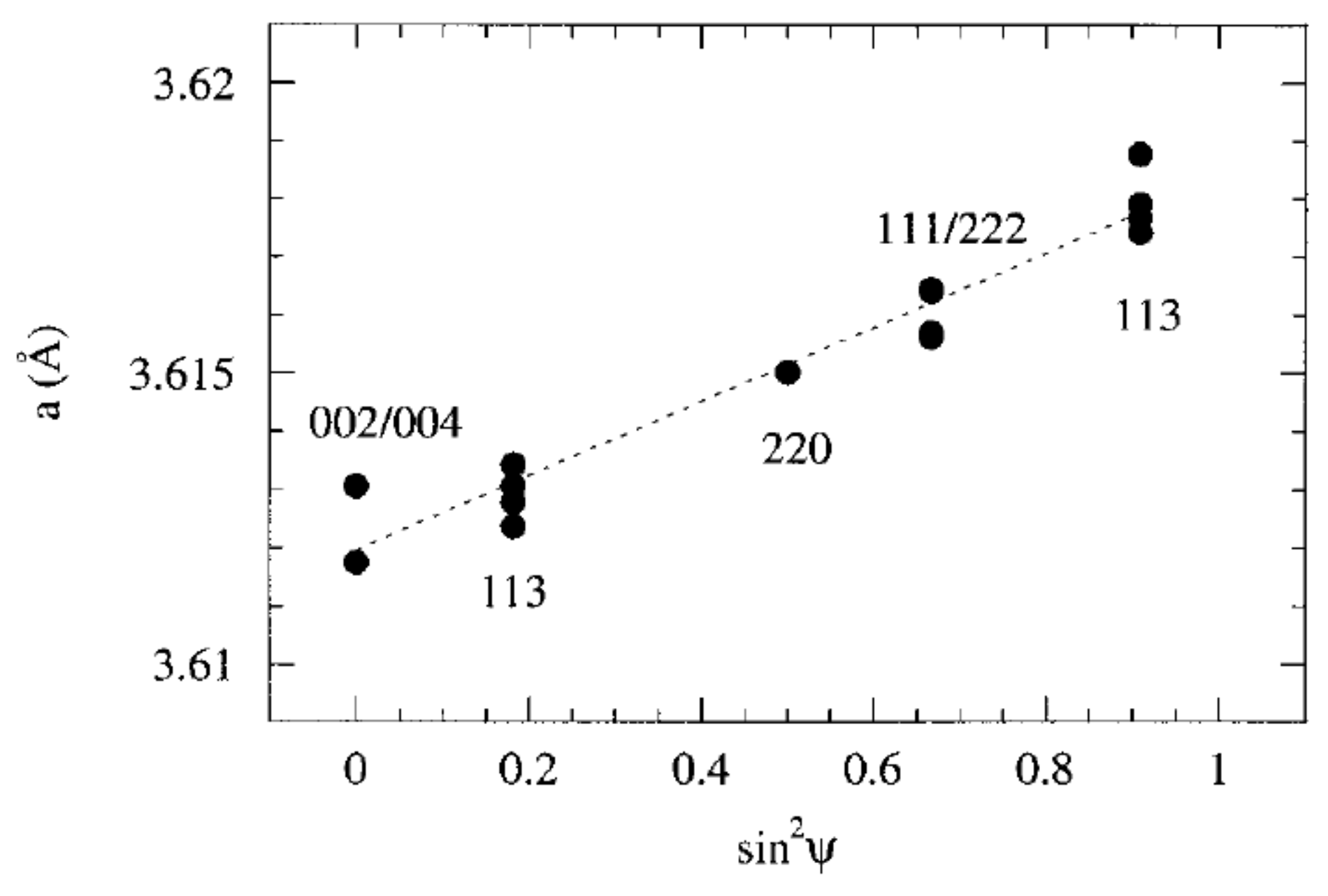
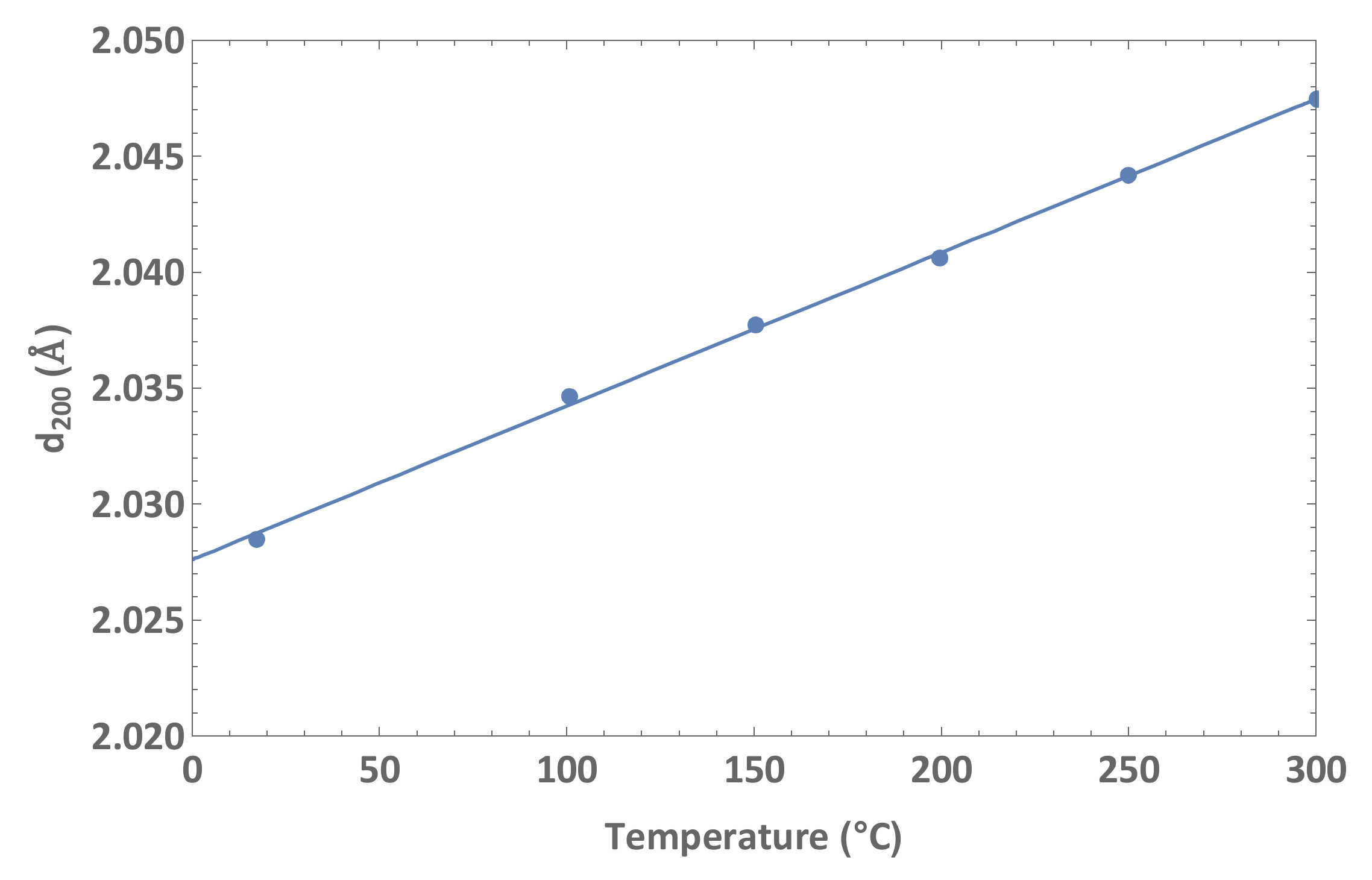
2.2. Analysis of Diffraction Peak Displacements
2.3. Diffraction Peak Shape Analysis
3. Recent Developments: Toward the 3D Imaging of Deformations
3.1. Focusing/Small Beams
3.1.1. Refractive Optics
3.1.2. Diffractive Optics
3.1.3. Focusing Mirrors
3.2. Scanning XRD Microscopy
3.3. Coherent Imaging by Diffraction under Bragg Conditions
3.4. Dark Field Imaging
4. Application Examples
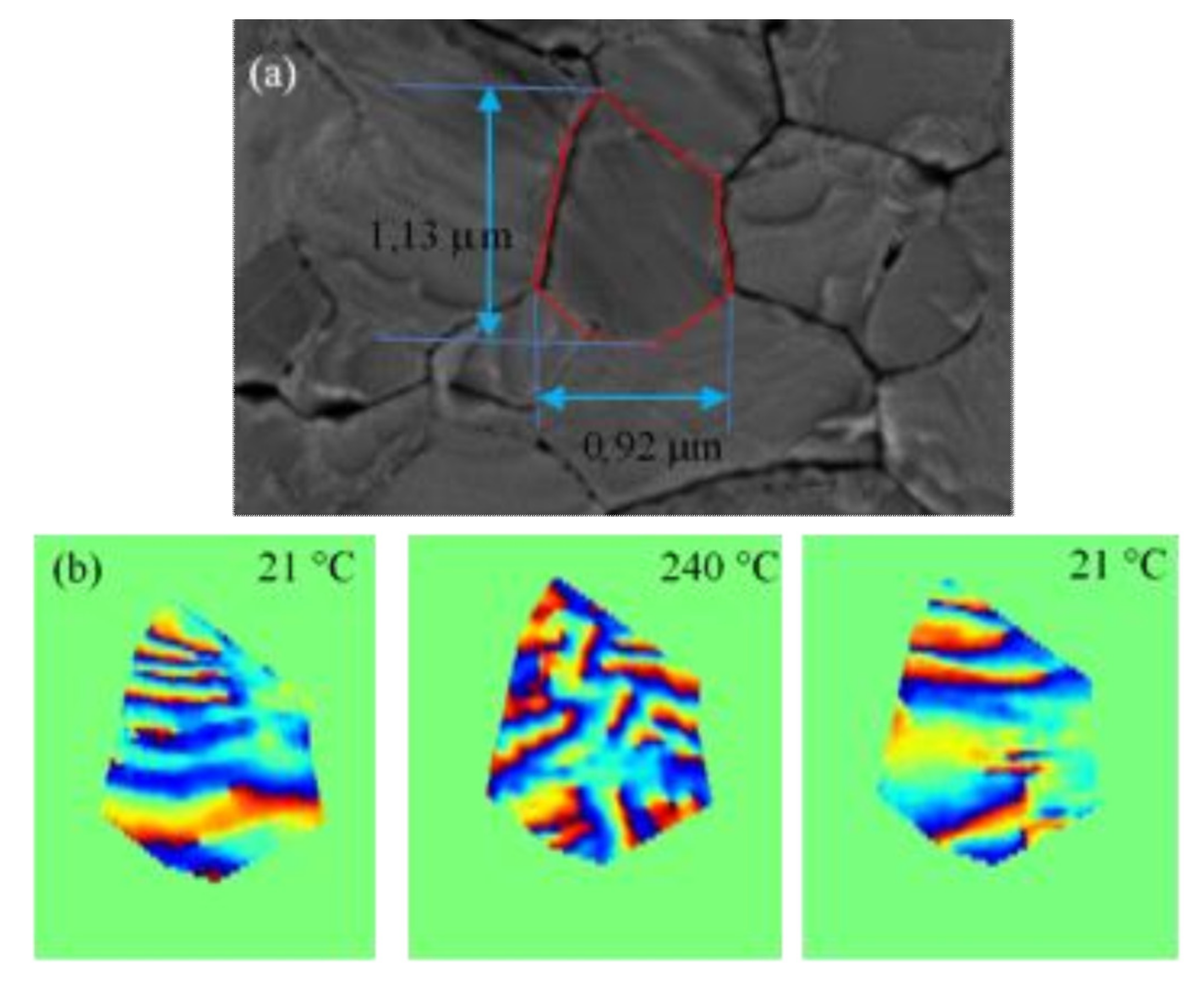
4.1. Coherent Bragg Imaging in Nanostructures
4.2. Imaging of Deformations by Scanning XRD Microscopy
4.3. Laue Micro-Diffraction Applied to the Mechanics of Nano and Micro-Objects
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thompson, S.E.; Armstrong, M.; Auth, C.; Cea, S.; Chau, R.; Glass, G.; Hoffman, T.; Klaus, J.; Ma, Z.; Mcintyre, B.; et al. A logic nanotechnology featuring strained-silicon. IEEE Electron. Device Lett. 2004, 25, 191–193. [Google Scholar] [CrossRef]
- Thomas, O.; Shen, Q.; Schieffer, P.; Tournerie, N.; Lepine, B. Interplay between anisotropic strain relaxation and uniaxial interface magnetic anisotropy in epitaxial Fe films on (001) GaAs. Phys. Rev. Lett. 2003, 90, 017205. [Google Scholar] [CrossRef] [Green Version]
- Signorello, G.; Karg, S.; Bjork, M.T.; Gotsmann, B.; Riel, H. Tuning the light emission from GaAs nanowires over 290 meV with uniaxial strain. Nano Lett. 2013, 13, 917–924. [Google Scholar] [CrossRef]
- Wang, H.; Xu, S.; Tsai, C.; Li, Y.; Liu, C.; Zhao, J.; Liu, Y.; Yuan, H.; Abild-Pedersen, F.; Prinz, F.B.; et al. Direct and continuous strain control of catalysts with tunable battery electrode materials. Science 2016, 354, 1031–1036. [Google Scholar] [CrossRef]
- Friedrich, W.; Knipping, P.; Laue, M. Interferenzerscheinungen bei Röntgenstrahlen. Sitz. Bayer. Akad. Wiss. 1913, 346, 971–988. [Google Scholar] [CrossRef] [Green Version]
- Aminoff, G. X-ray asterism on Laue-photogramms. Geol. Föreningen I Stockh. Förhandlingar 1919, 41, 534–538. [Google Scholar] [CrossRef]
- Czochralski, J. Moderne Metallkunde; Julius Springer: Berlin, Germany, 1924. [Google Scholar]
- Taylor, G.I. The mechanism of plastic deformation of crystals, Part I–Theoretical. Proc. Roy. Soc. London A 1934, 145, 362–387. [Google Scholar]
- Polanyi, M. Über eine Art Gitterstörung, die einen Kristall plastisch machen könnte. Z. Für Phys. 1934, 89, 660–664. [Google Scholar] [CrossRef]
- Orowan, E. Zur Kristallplastizität. I—Tieftemperaturplastizität und Beckersche Formel. Z. Für Phys. 1934, 89, 605–613. [Google Scholar] [CrossRef]
- Orowan, E. Zur Kristallplastizität. II—Die dynamische Auffassung der Kristallplastizität. Z. Für Phys. 1934, 89, 614–633. [Google Scholar] [CrossRef]
- Orowan, E. Zur Kristallplastizität. III—Über den Mechanismus des Gleitvorganges. Z. Für Phys. 1934, 89, 634–659. [Google Scholar] [CrossRef]
- Lester, H.; Aborn, R. Behavior under stress of the iron crystals. Army Ordnance 1925, 6, 120. [Google Scholar]
- Glocket, R.; Osswaldn, E. Unique determination of the principal stresses with X-rays. Z. Tech. Phys. 1935, 161, 237. [Google Scholar]
- Noyan, I.C.; Cohen, J.B. Residual Stress: Measurement by Diffraction and Interpretation; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Spolenak, R.; Brown, W.L.; Tamura, N.; MacDowell, A.A.; Celestre, R.S.; Padmore, H.A.; Valek, B.; Bravman, J.C.; Marieb, T.; Fujimoto, H.; et al. Local plasticity of Al thin films as revealed by X-ray microdiffraction. Phys. Rev. Lett. 2003, 90, 096102. [Google Scholar] [CrossRef]
- Chahine, G.A.; Richard, M.-I.; Homs-Regojo, R.A.; Tran-Caliste, T.N.; Carbone, D.; Jacques, V.L.R.; Grifone, R.; Boesecke, P.; Katzer, J.; Costina, I.; et al. Imaging of strain and lattice orientation by quick scanning X-ray microscopy combined with three-dimensional reciprocal space mapping. J. Appl. Crystallogr. 2014, 47, 762–769. [Google Scholar] [CrossRef]
- Simons, H.; King, A.; Ludwig, W.; Detlefs, C.; Pantleon, W.; Schmidt, S.; Stohr, F.; Snigireva, I.; Snigirev, A.; Poulsen, H.F. Dark-field X-ray microscopy for multiscale structural characterization. Nat. Comm. 2015, 6, 6098. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, M.A.; Williams, G.J.; Vartanyants, I.A.; Harder, R.; Robinson, I.K. Three-dimensional mapping of a deformation field inside a nanocrystal. Nature 2006, 442, 63. [Google Scholar] [CrossRef]
- Robinson, I.; Harder, R. Coherent X-ray diffraction imaging of strain at the nanoscale. Nature Mat. 2009, 8, 291–298. [Google Scholar] [CrossRef]
- Berar, J.F.; Boudet, N.; Breugnon, P.; Caillot, B.; Chantepie, B.; Clemens, J.C.; Delpierre, P.; Dinkespiller, B.; Godiot, S.; Meessen, C.; et al. XPAD3 hybrid pixel detector applications. Nucl. Instr. Meth. Phys. Res. A 2009, 607, 233–235. [Google Scholar] [CrossRef]
- Henrich, B.; Bergamaschi, A.; Broennimann, C.; Dinapoli, R.; Eikenberry, E.F.; Johnson, I.; Kobas, M.; Kraft, P.; Mozzanica, A.; Schmitt, B. PILATUS: A single photon counting pixel detector for X-ray applications. Nucl. Instr. Meth. Phys. Res. A 2009, 607, 247–249. [Google Scholar] [CrossRef]
- Ponchut, C.; Rigal, J.M.; Clément, J.; Papillon, E.; Homs, A.; Petitdemange, S. MAXIPIX, a fast readout photon-counting X-ray area detector for synchrotron applications. J. Instrum. 2001, 6, C01069. [Google Scholar] [CrossRef] [Green Version]
- Warren, B. X-ray Diffraction; Dover: New York, USA, 1991. [Google Scholar]
- Cowley, J. Diffraction Physics, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- Authier, A. Dynamical Theory of X-ray Diffraction; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Busing, W.R.; Levyn, H.A. Angle calculations for 3- and 4- circle X-ray and neutron diffractometers. Acta Crystallogr. 1967, 22, 467. [Google Scholar] [CrossRef]
- Patterson, A.L. International Tables of Crystallography; International Union of Crystallography: Chester, UK, 1959; Volume 2. [Google Scholar]
- Giacovazzo, C. Fundamentals of Crystallography; IUCr: Chester, UK; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Kleinman, L. Deformation potentials in silicon: I. Uniaxial strain. Phys. Rev. 1962, 128, 2614–2621. [Google Scholar] [CrossRef]
- Segmüller, A. Observation of bond-bending in strained germanium. Phys. Lett. 1963, 4, 277–278. [Google Scholar] [CrossRef]
- Labat, S.; Gergaud, P.; Thomas, O.; Gilles, B.; Marty, A. Interdependance between elastic strain and segregation in metallic multilayers: An X-ray diffraction study of (111) Au/Ni multilayers. J. Appl. Phys. 2000, 87, 1172–1181. [Google Scholar] [CrossRef]
- Piot, A. Etude de la fabrication et de la transduction d’un microgyromètre piézoélectrique tri-axial en GaAs. Ph.D. Thesis, Université Paris-Saclay, Paris, France, 2018. [Google Scholar]
- Scherrer, P. Bestimmung der Grosse und der Inneren Struktur von Kolloidteilchen Mittels Rontgenstrahlen. Nachrichten von der Gesellschaft der Wissenschaften, Gottingen. Math. Phys. Kl. 1918, 2, 98–100. [Google Scholar]
- Averbach, B.L.; Warren, B.E. Interpretation of X-ray patterns of cold-worked metal. J. Appl. Phys. 1949, 20, 885–886. [Google Scholar] [CrossRef]
- Wilson, A.J.C. X-ray Optics; Methuen: London, UK, 1962. [Google Scholar]
- Groma, I. X-ray line broadening due to an inhomogeneous dislocation distribution. Phys. Rev. B 1998, 57, 7535. [Google Scholar] [CrossRef]
- Mittemeijer, E.; Scardi, P. Diffraction Analysis of the Microstructure of Materials; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Levine, H.; Gantil, P.; Larson, B.; Tischler, J.; Kassner, M.; Liu, W. Validating classical line profile analyses using microbeam diffraction from individual dislocation cell walls and cell interiors. J. Appl. Crystallogr. 2012, 45, 157–165. [Google Scholar] [CrossRef]
- Mughrabi, H. Dislocation wall and cell structures and long-range internal stresses in deformed metal crystals. Acta Metall. 1983, 31, 1367–1379. [Google Scholar] [CrossRef]
- Hofmann, F.; Abbey, B.; Liu, W.; Xu, R.; Usher, B.F.; Balaur, E.; Liu, Y. X-ray micro-beam characterization of lattice rotations and distortions due to an individual dislocation. Nat. Com. 2013, 4, 2774. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Charalambous, P.; Kirz, J.; Sayre, D. Extending the methodology of X-ray crystallography to allow imaging of micrometer-sized non-crystalline specimens. Nature 1999, 400, 342–344. [Google Scholar] [CrossRef]
- Robinson, I.K.; Vartanyants, I.A.; Williams, G.J.; Pfeifer, M.A.; Pitney, J.A. Reconstruction of the shapes of gold nanocrystals using coherent X-ray diffraction. Phys. Rev. Lett. 2001, 87, 195505. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S. A dynamical theory of diffraction by a distorted crystal. J. Phys. Soc. Jpn. 1969, 26, 1239–1253. [Google Scholar] [CrossRef]
- Jacques, V.L.R.; Ravy, S.; LeBolloc’h, D.; Pinsolle, E.; Sauvage-Simkin, M.; Livet, F. Bulk dislocation core dissociation probed by coherent X-rays in silicon. Phys. Rev. Lett. 2011, 106, 065502. [Google Scholar] [CrossRef]
- Pizzagalli, L.; Rabier, J.; Godet, J.; Devincre, B.; Kubin, L. Comment on “Bulk Dislocation Core Dissociation Probed by Coherent X rays in Silicon”. Phys. Rev. Lett. 2011, 107, 199601. [Google Scholar] [CrossRef]
- Jacques, V.L.R.; Ravy, S.; LeBolloc’h, D.; Pinsolle, E.; Sauvage-Simkin, M.; Livet, F. Comment on “Bulk Dislocation Core Dissociation Probed by Coherent X Rays in Silicon” Reply. Phys. Rev. Lett. 2011, 107, 199602. [Google Scholar] [CrossRef]
- Takahashi, Y.; Suzuki, A.; Furutaku, S.; Yamauchi, K.; Kohmura, Y.; Ishikawa, T. Bragg X-ray ptychography of a silicon crystal: Visualization of the dislocation strain field and the production of a vortex beam. Phys. Rev. B 2013, 87, 121201. [Google Scholar] [CrossRef]
- Ulvestad, A.; Singer, A.; Clark, J.N.; Cho, H.M.; Kim, J.W.; Harder, R.; Maser, J.; Meng, Y.S.; Shpyrko, O.G. Topological defect dynamics in operando battery nanoparticles. Science 2015, 348, 1344–1347. [Google Scholar] [CrossRef] [Green Version]
- Ulvestad, A.; Welland, M.J.; Cha, W.; Liu, Y.; Kim, J.W.; Harder, R.; Maxey, E.; Clark, J.N.; Highland, M.J.; You, H.; et al. Three-dimensional imaging of dislocation dynamics during the hydriding phase transformation. Nat. Mat. 2017, 16, 565–571. [Google Scholar] [CrossRef]
- Dupraz, M.; Beutier, G.; Cornelius, T.W.; Parry, G.; Ren, Z.; Labat, S.; Richard, M.-I.; Chahine, G.A.; Kovalenko, O.; Rabkin, E.; et al. 3D imaging of a dislocation loop at the onset of plasticity in an indented nanocrystal. Nano Lett. 2017, 17, 6696. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Phillips, N.W.; Das, S.; Karamched, P.; Hughes, G.M.; Douglas, J.O.; Cha, W.; Liu, W. Nanoscale imaging of the full strain tensor of specific dislocations extracted from a bulk sample. Phys. Rev. Mat. 2020, 4, 013801. [Google Scholar] [CrossRef] [Green Version]
- Snigirev, A.; Kohn, V.; Snigireva, I.; Lengeler, B. A compound refractive lens for focusing high-energy X-rays. Nature 1996, 384, 49–51. [Google Scholar] [CrossRef]
- Lengeler, B.; Schroer, C.; Tümmler, J.; Benner, B.; Richwin, M.; Snigirev, A.; Snigireva, I.; Drakopoulos, M. Imaging by parabolic refractive lenses in the hard X-ray range. J. Synchrotron Radiat. 1999, 6, 1153–1167. [Google Scholar] [CrossRef]
- Kirz, J. Phase Zone Plates for X-rays and Extreme UV. J. Opt. Soc. Am. 1974, 64, 301–309. [Google Scholar] [CrossRef]
- Jefimovs, K.; Bunk, O.; Pfeiffer, F.; Grolimund, D.; van der Veen, J.F.; David, C. Fabrication of Fresnel zone plates for hard X-rays. Microelectron. Eng. 2007, 84, 1467–1470. [Google Scholar] [CrossRef]
- Kang, H.C.; Maser, J.; Stephenson, G.B.; Liu, C.; Conley, R.; Macrander, A.T.; Vogt, S. Nanometer linear focusing of hard x rays by a multilayer Laue lens. Phys. Rev. Lett. 2006, 96, 127401. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yan, H.; Nazaretski, E.; Conley, R.; Bouet, N.; Zhou, J.; Lauer, K.; Li, L.; Eom, D.; Legnini, D.; et al. 11 nm hard X-ray focus from a large-aperture multilayer Laue lens. Sci. Rep. 2013, 3, 3562. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, P.; Baez, A.V. Formation of optical images by X-rays. J. Opt. Soc. Am. 1948, 38, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Hignette, O.; Cloetens, P.; Rostaing, G.; Bernard, P.; Morawe, C. Efficient sub 100 nm focusing of hard X rays. Rev. Sci. Instrum. 2005, 76, 063709. [Google Scholar] [CrossRef]
- Riekel, C.; Burghammer, M.; Müller, M. Microbeam small-angle scattering experiments and their combination with microdiffraction. J. Appl. Crystallogr. 2000, 33, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Manceau, A.; Tamura, N.; Marcus, N.; MacDowell, A.A.; Celestre, R.S.; Sublett, R.E.; Sposito, G.; Padmore, H.A. Deciphering Ni sequestration in soil ferromanganese nodules by combining X-ray fluorescence, absorption, and diffraction at micrometer scales of resolution. Am. Mineral. 2002, 87, 1494–1499. [Google Scholar] [CrossRef]
- Tamura, N.; MacDowell, A.A.; Spolenak, R.; Valek, B.; Bravman, J.C.; Brown, W.L.; Celestre, R.S.; Padmore, H.A.; Batterman, B.W.; Patel, J.R. Scanning X-ray microdiffraction with submicrometer white beam for strain/stress and orientation mapping in thin films. J. Synchrotron Radiat. 2003, 10, 137–143. [Google Scholar] [CrossRef]
- Send, S.; Abboud, A.; Leitenberger, W.; Weiss, M.S.; Hartmann, R.; Strüder, L.; Pietsch, U. Analysis of polycrystallinity in hen egg-white lysozyme using a pnCCD. J. Appl. Crystallogr. 2012, 45, 517–522. [Google Scholar] [CrossRef]
- Robach, O.; Micha, J.-S.; Ulrich, O.; Geaymond, O.; Sicardy, O.; Hartwig, J.; Rieutord, F. A tunable multicolor ‘rainbow’ filter for improved stress and dislocation density field mapping in polycrystals using X-ray Laue microdiffraction. Acta Crystallogr. A 2013, 69, 164–170. [Google Scholar] [CrossRef]
- Larson, B.; Yang, W.; Ice, G.; Budai, J.; Tischler, J. Three-dimensional X-ray structural microscopy with submicrometre resolution. Nature 2002, 415, 887–890. [Google Scholar] [CrossRef]
- Richard, M.I.; Cornelius, T.W.; Lauraux, F.; Molin, J.B.; Kirchlechner, C.; Leake, S.J.; Carnis, J.; Schülli, T.U.; Thilly, L.; Thomas, O. Variable-Wavelength Quick Scanning Nanofocused X-ray Microscopy for In Situ Strain and Tilt Mapping. Small 2020, 16, 1905990. [Google Scholar] [CrossRef]
- Leclere, C.; Cornelius, T.W.; Ren, Z.; Robach, O.; Micha, J.-S.; Davydok, A.; Ulrich, O.; Richter, G.; Thomas, O. KB scanning of X-ray beam for Laue microdiffraction on accelero-phobic samples: Application to in situ mechanically loaded nanowires. J. Synchrotron Radiat. 2016, 23, 1395–1400. [Google Scholar] [CrossRef]
- Sayre, D. Some implications of a theorem due to Shannon. Acta Crystallogr. 1952, 5, 843. [Google Scholar] [CrossRef] [Green Version]
- Born, M.; Wolf, E. Principles of Optics; Pergamon Press: Oxford, UK, 1980. [Google Scholar]
- Gerchberg, R.W.; Saxton, W.O. A practical algorithm for the determination of the phase from image and diffraction plane pictures. Optik 1972, 35, 237. [Google Scholar]
- Fienup, J.R. Reconstruction of an object from the modulus of its Fourier transform. Opt. Lett. 1978, 3, 27–29. [Google Scholar] [CrossRef]
- Clark, J.N.; Huang, X.; Harder, R.; Robinson, I.K. High-resolution three-dimensional partially coherent diffraction imaging. Nat. Commun 2012, 3, 993. [Google Scholar] [CrossRef]
- Harder, R.; Pfeifer, M.A.; Williams, G.J.; Vartaniants, I.A.; Robinson, I.K. Orientation variation of surface strain. Phys. Rev. B 2007, 76, 115425. [Google Scholar] [CrossRef] [Green Version]
- Mandula, O.; Elzo Aizarna, M.; Eymery, J.; Burghammer, M.; Favre-Nicolin, V. PyNX.Ptycho: A computing library for X-ray coherent diffraction imaging of nanostructures. J. Appl. Crystallogr. 2016, 49, 1842–1848. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Girard, G.; Leake, S.; Carnis, J.; Chushkin, Y.; Kieffer, J.; Paleo, P.; Richard, M.-I. PyNX: High-performance computing toolkit for coherent X-ray imaging based on operators. J. Appl. Crystallogr. 2020, 53, 1404–1413. [Google Scholar] [CrossRef]
- Chapman, H.N.; Barty, A.; Marchesini, S.; Noy, A.; Hau-Riege, S.R.; Cui, C.; Howells, M.R.; Rosen, R.; He, H.; Spence, J.C.H.; et al. High-resolution ab initio three-dimensional X-ray diffraction microscopy. J. Opt. Soc. Am. A 2006, 23, 1179–1200.B82. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Leake, S.; Chushkin, Y. Free log-likelihood as an unbiased metric for coherent diffraction imaging. Sci. Rep. 2020, 10, 2664. [Google Scholar] [CrossRef]
- Labat, S.; Richard, M.I.; Dupraz, M.; Gailhanou, M.; Beutier, G.; Verdier, M.; Mastropietro, F.; Cornelius, T.W.; Schülli, T.U.; Eymery, J.; et al. Inversion Domain Boundaries in GaN Wires Revealed by Coherent Bragg Imaging. ACS Nano 2015, 9, 9210–9216. [Google Scholar] [CrossRef]
- Fernandez, S.; Gao, L.; Hofmann, J.P.; Carnis, J.; Labat, S.; Chahine, G.; van Hoof, A.J.F.; Verhoeven, M.W.G.M.; Favre-Nicolin, V.; Schülli, T.; et al. In situ structural evolution of single particle model catalysts under ambient pressure reaction conditions. Nanoscale 2019, 11, 331. [Google Scholar] [CrossRef]
- Kim, D.; Chung, M.; Carnis, J.; Kim, S.; Yun, K.; Kang, J.; Cha, W.; Cherukara, M.J.; Maxey, E.; Harder, R.; et al. Active site localization of methane oxidation on Pt nanocrystals. Nat. Com. 2018, 9, 3422. [Google Scholar] [CrossRef]
- Vaxelaire, N.; Labat, S.; Cornelius, T.; Kirchlechner, C.; Keckes, J.; Schulli, T.; Thomas, O. New insights into single-grain mechanical behavior from temperature-dependent 3-D coherent X-ray diffraction. Acta Mater. 2014, 78, 46–55. [Google Scholar] [CrossRef]
- Yau, A.; Cha, W.; Kanan, M.W.; Stephenson, G.B.; Ulvestad, A. Bragg coherent diffractive imaging of single-grain defect dynamics in polycrystalline films. Science 2017, 356, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, W. Beugung im inhomogenen Primärstrahlwellenfeld. I. Prinzip einer Phasenmessung von Elektronenbeungungsinterferenzen. Acta Crystallogr. A 1969, 25, 495–501. [Google Scholar] [CrossRef]
- Rodenburg, J.M.; Bates, R.H.T. The theory of super-resolution electron microscopy via Wigner-distribution deconvolution. Philos. Trans. R. Soc. Lond. Ser. A 1992, 339, 521–553. [Google Scholar]
- Thibault, P.; Dierolf, M.; Menzel, A.; Bunk, O.; David, C.; Pfeiffer, F. High-resolution Scanning X-ray Diffraction Microscopy. Science 2008, 321, 379–382. [Google Scholar] [CrossRef]
- Diaz, A.; Trtik, P.; Guizar-Sicairos, M.; Menzel, A.; Thibault, P.; Bunk, O. Quantitative X-ray phase nanotomography. Phys. Rev. B 2012, 85, 020104. [Google Scholar] [CrossRef] [Green Version]
- Holler, M.; Guizar-Sicairos, M.; Tsai, E.H.R.; Dinapoli, R.; Müller, E.; Bunk, O.; Raabe, J.; Aeppli, G. High-resolution non-destructive three-dimensional imaging of integrated circuits. Nature 2017, 543, 402–406. [Google Scholar] [CrossRef]
- Godard, P.; Carbone, G.; Allain, M.; Mastropietro, F.; Chen, G.; Capello, L.; Diaz, A.; Metzger, T.H.; Stangl, J.; Chamard, V. Three-dimensional high-resolution quantitative microscopy of extended crystals. Nat. Comm. 2011, 2, 568. [Google Scholar] [CrossRef]
- Lengeler, B.; Schroer, C.G.; Richwin, M.; Tümmler, J.; Drakopoulos, M.; Snigirev, A.; Snigireva, I. A microscope for hard X-rays based on parabolic compound refractive lenses. Appl. Phys. Lett. 1999, 74, 3924–3926. [Google Scholar] [CrossRef] [Green Version]
- Simons, H.; Haugen, A.B.; Jakobsen, A.C.; Schmidt, S.; Stöhr, F.; Majkut, M.; Detlefs, C.; Daniels, J.; Damjanovic, D.; Poulsen, H.F. Long-range symmetry breaking in embedded ferroelectrics. Nat. Mater. 2018, 17, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Newton, M.C.; Leake, S.; Harder, R.; Robinson, I.K. Three-dimensional imaging of strain in a single ZnO nanorod. Nat. Mat. 2010, 9, 120–124. [Google Scholar] [CrossRef]
- Lancon, F.; Genovese, L.; Eymery, J. Towards simulation at picometer-scale resolution: Revisiting inversion domain boundaries in GaN. Phys. Rev. B 2018, 98, 165306. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.N.; Beitra, L.; Xiong, G.; Higginbotham, A.; Fritz, D.M.; Lemke, H.T.; Zhu, D.; Chollet, M.; Williams, G.J.; Messerschmidt, M.; et al. Ultrafast Three-Dimensional Imaging of Lattice Dynamics in Individual Gold Nanocrystals. Science 2013, 341, 56. [Google Scholar] [CrossRef] [Green Version]
- Vianne, B.; Richard, M.-I.; Escoubas, S.; Labat, S.; Schülli, T.; Chahine, G.; Fiori, V.; Thomas, O. Through-silicon via-induced strain distribution in silicon interposer. Appl. Phys. Lett. 2015, 106, 141905. [Google Scholar] [CrossRef]
- Vianne, B.; Escoubas, S.; Richard, M.-I.; Labat, S.; Chahine, G.; Schulli, T.; Farcy, A.; Bar, P.; Fiori, V.; Thomas, O. Strain and tilt mapping in silicon around copper filled TSVs using advanced X-ray nano-diffraction. Microelectron. Eng. 2015, 137, 117–123. [Google Scholar] [CrossRef]
- Escoubas, S.; Vianne, B.; Richard, M.-I.; Farcy, A.; Fiori, V.; Thomas, O. Strain Distribution Induced in SOI Photonic Substrate by Through Silicon via Using Advanced Scanning X-ray Nano-Diffraction. IEEE Trans. Device Mater. Reliab. 2018, 18, 529–533. [Google Scholar] [CrossRef]
- Mondiali, V.; Bollani, M.; Cecchi, S.; Richard, M.-I.; Schülli, T.; Chahine, G.; Chrastina, D. Dislocation engineering in SiGe on periodic and aperiodic Si(001) templates studied by fast scanning X-ray nanodiffraction. Appl. Phys. Lett. 2014, 104, 021918. [Google Scholar] [CrossRef] [Green Version]
- Mondiali, V.; Bollani, M.; Chrastina, D.; Rubert, R.; Chahine, G.; Richard, M.I.; Cecchi, S.; Gagliano, L.; Bonera, E.; Schülli, T.; et al. Strain release management in SiGe/Si films by substrate patterning. Appl. Phys. Lett. 2014, 105, 242103. [Google Scholar] [CrossRef] [Green Version]
- Zoellner, M.H.; Richard, M.-I.; Chahine, G.A.; Zaumseil, P.; Reich, C.; Capellini, G.; Montalenti, F.; Marzegalli, A.; Xie, Y.-H.; Schülli, T.U.; et al. Imaging Structure and Composition Homogeneity of 300 mm SiGe Virtual Substrates for Advanced CMOS Applications by Scanning X-ray Diffraction Microscopy. ACS Appl. Mater. Interfaces 2015, 7, 9031–9037. [Google Scholar] [CrossRef] [Green Version]
- Zoellner, M.H.; Chahine, G.A.; Lahourcade, L.; Mounir, C.; Manganelli, C.L.; Schülli, T.U.; Schwarz, U.T.; Zeisel, R.; Schroeder, T. Correlation of Optical, Structural, and Compositional Properties with V-Pit Distribution in InGaN/GaN Multiquantum Wells. ACS Appl. Mater. Interfaces 2019, 11, 22834–22839. [Google Scholar] [CrossRef]
- Etzelstorfer, T.; Süess, M.J.; Schiefler, G.L.; Jacques, V.L.R.; Carbone, D.; Chrastina, D.; Isella, G.; Spolenak, R.; Stangl, J.; Sigg, H.; et al. Scanning X-ray strain microscopy of inhomogeneously strained Ge micro-bridges. J. Synchrotron Radiat. 2014, 21, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Chahine, G.A.; Zoellner, M.H.; Richard, M.-I.; Guha, S.; Reich, C.; Zaumseil, P.; Capellini, G.; Schroeder, T.; Schülli, T.U. Strain and lattice orientation distribution in SiN/Ge complementary metal–oxide–semiconductor compatible light emitting microstructures by quick X-ray nano-diffraction microscopy. Appl. Phys. Lett. 2015, 106, 071902. [Google Scholar] [CrossRef]
- Evans, P.G.; Savage, D.E.; Prance, J.R.; Simmons, C.B.; Lagally, M.G.; Coppersmith, S.N.; Eriksson, M.A.; Schülli, T.U. Nanoscale Distortions of Si Quantum Wells in Si/SiGe Quantum-Electronic Heterostructures. Adv. Mater. 2012, 24, 5217–5221. [Google Scholar] [CrossRef]
- Ristanović, Z.; Hofmann, J.P.; Richard, M.-I.; Jiang, T.; Chahine, G.A.; Schülli, T.U.; Meirer, F.; Weckhuysen, B.M. X-ray Excited Optical Fluorescence and Diffraction Imaging of Reactivity and Crystallinity in a Zeolite Crystal: Crystallography and Molecular Spectroscopy in One. Angew. Chem. Int. Ed. 2016, 55, 7496–7500. [Google Scholar] [CrossRef] [Green Version]
- Filippelli, E.; Chahine, G.; Borbély, A. Evaluation of intragranular strain and average dislocation density in single grains of a polycrystal using K-map scanning. J. Appl. Crystallogr. 2016, 49, 1814–1817. [Google Scholar] [CrossRef]
- Paskiewicz, D.M.; Savage, D.E.; Holt, M.V.; Evans, P.G.; Lagally, M.G. Nanomembrane-based materials for Group IV semiconductor quantum electronics. Sci. Rep. 2015, 4, 4218. [Google Scholar] [CrossRef] [Green Version]
- Marçal, L.A.B.; Richard, M.-I.; Magalhães-Paniago, R.; Cavallo, F.; Lagally, M.G.; Schmidt, O.G.; Schülli, T.Ü.; Deneke, C.; Malachias, A. Direct evidence of strain transfer for InAs island growth on compliant Si substrates. Appl. Phys. Lett. 2015, 106, 151905. [Google Scholar] [CrossRef]
- Highland, M.J.; Hruszkewycz, S.O.; Fong, D.D.; Thompson, C.; Fuoss, P.H.; Calvo-Almazan, I.; Maddali, S.; Ulvestad, A.; Nazaretski, E.; Huang, X.; et al. In-situ synchrotron X-ray studies of the microstructure and stability of In2O3 epitaxial films. Appl. Phys. Lett. 2017, 111, 161602. [Google Scholar] [CrossRef]
- Richard, M.-I.; Zoellner, M.H.; Chahine, G.A.; Zaumseil, P.; Capellini, G.; Häberlen, M.; Storck, P.; Schülli, T.U.; Schroeder, T. Structural Mapping of Functional Ge Layers Grown on Graded SiGe Buffers for sub-10 nm CMOS Applications Using Advanced X-ray Nanodiffraction. ACS Appl. Mater. Interfaces 2015, 7, 26696. [Google Scholar] [CrossRef]
- Johannes, A.; Rensberg, J.; Grünewald, T.A.; Schöppe, P.M.; Rosenthal, M.; Ronning, C.; Burghammer, M. Determination of the full deformation tensor by multi-Bragg fast scanning nano X-ray diffraction. J. Appl. Crystallogr. 2020, 53, 99–106. [Google Scholar] [CrossRef]
- Xu, W.; Xu, W.; Bouet, N.; Zhou, J.; Yan, H.; Huang, X.; Pattammattel, A.; Gao, Y.; Lu, M.; Zalalutdinov, M.K.; et al. 2D MEMS-based multilayer Laue lens nanofocusing optics for high-resolution hard X-ray microscopy. Opt. Express 2020, 28, 17660. [Google Scholar] [CrossRef]
- Bajt, S.; Prasciolu, M.; Fleckenstein, H.; Domaracký, M.; Chapman, H.N.; Morgan, A.J.; Yefanov, O.; Messerschmidt, M.; Du, Y.; Murray, K.T.; et al. X-ray focusing with efficient high-NA multilayer Laue lenses. Light: Sci. Appl. 2018, 7, 17162. [Google Scholar] [CrossRef]
- Nazaretski, E.; Yan, H.; Lauer, K.; Bouet, N.; Huang, X.; Xu, W.; Zhou, J.; Shu, D.; Hwu, Y.; Chu, Y.S. Design and performance of an X-ray scanning microscope at the Hard X-ray Nanoprobe beamline of NSLS-II. J. Synchrotron Radiat. 2017, 24, 1113–1119. [Google Scholar] [CrossRef]
- Winarski, R.P.; Holt, M.V.; Rose, V.; Fuesz, P.; Carbaugh, D.; Benson, C.; Shu, D.; Kline, D.; Stephenson, G.B.; McNulty, I.; et al. A hard X-ray nanoprobe beamline for nanoscale microscopy. J. Synchrotron Radiat. 2012, 19, 1056–1060. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Bouet, N.; Zhou, J.; Huang, X.; Nazaretski, E.; Xu, W.; Cocco, A.P.; Chiu, W.K.S.; Brinkman, K.S.; Chu, Y.S. Multimodal hard X-ray imaging with resolution approaching 10 nm for studies in material science. Nano Futures 2018, 2, 011001. [Google Scholar] [CrossRef]
- Medjoubi, K.; Thompson, A.; Bérar, J.-F.; Clemens, J.-C.; Delpierre, P.; Da Silva, P.; Dinkespiler, B.; Fourme, R.; Gourhant, P.; Guimaraes, B.; et al. Energy resolution of the CdTe-XPAD detector: Calibration and potential for Laue diffraction measurements on protein crystals. J. Synchrotron Radiat. 2012, 19, 323–331. [Google Scholar] [CrossRef]
- Mocuta, C.; Richard, M.-I.; Fouet, J.; Stanescu, S.; Barbier, A.; Guichet, C.; Thomas, O.; Hustache, S.; Zozulya, A.; Thiaudiere, D. Fast pole figure acquisition using area detectors at the DiffAbs beamline—Synchrotron SOLEIL. J. Appl. Crystallogr. 2013, 46, 1842–1853. [Google Scholar] [CrossRef] [Green Version]
- Maaß, R.; Van Petegem, S.; Van Swygenhoven, H.; Derlet, P.M.; Volkert, C.A.; Grolimund, D. Time-resolved Laue diffraction of deforming micropillars. Phys. Rev. Lett. 2007, 99, 145505. [Google Scholar] [CrossRef] [Green Version]
- Van Swygenhoven, H.; Van Petegem, S. The use of Laue microdiffraction to study small-scale plasticity. JOM 2010, 62, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Kirchlechner, C.; Keckes, J.; Micha, J.-S.; Dehm, G. In situ µLaue: Instrumental setup for the deformation of micron sized samples. Adv. Eng. Mat. 2011, 13, 837–844. [Google Scholar] [CrossRef]
- Ren, Z.; Mastropietro, F.; Langlais, S.; Davydok, A.; Richard, M.-I.; Thomas, O.; Dupraz, M.; Verdier, M.; Beutier, G.; Boesecke, P.; et al. Scanning force microscopy for in situ nanofocused X-ray diffraction studies. J. Synchrotron Radiat. 2014, 21, 1128–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchlechner, C.; Keckes, J.; Motz, C.; Grosinger, W.; Kapp, M.W.; Micha, J.S.; Ulrich, O.; Dehm, G. Impact of instrumental constraints and imperfections on the dislocation structure in micron-sized Cu compression pillars. Acta Mater. 2011, 59, 5618–5626. [Google Scholar] [CrossRef]
- Kirchlechner, C.; Imrich, P.; Grosinger, W.J.; Kapp, M.W.; Keckes, J.; Micha, J.S.; Ulrich, O.; Thomas, O.; Labat, S.; Motz, C.S.; et al. Expected and unexpected plastic behavior at the micron scale: An in situ micro-Laue tensile study. Acta Mater. 2012, 60, 1252–1258. [Google Scholar] [CrossRef]
- Robach, O.; Kirchlechner, C.; Micha, J.S.; Ulrich, O.; Biquard, X.; Geaymond, O.; Castelnau, O.; Bornert, M.; Petit, J.; Sicardy, O.; et al. Laue Microdiffraction at the ESRF. In Strain and Dislocation Gradients from Diffraction; Barabash, R., Ice, G., Eds.; Imperial College Press: London, UK, 2014; pp. 156–204. [Google Scholar]
- Maaß, R.; Van Petegem, S.; Ma, D.; Zimmermann, J.; Grolimund, D.; Roters, F.; Van Swygenhoven, H.; Raabe, D. Smaller is stronger: The effect of strain hardening. Acta Mater. 2009, 57, 5996–6005. [Google Scholar] [CrossRef]
- Leclere, C.; Cornelius, T.W.; Ren, Z.; Davydok, A.; Micha, J.-S.; Robach, O.; Richter, G.; Belliard, L.; Thomas, O. In-situ bending of an Au nanowire monitored by micro Laue diffraction. J. Appl. Crystallogr. 2015, 48, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Cornelius, T.W.; Leclere, C.; Davydok, A.; Micha, J.-S.; Robach, O.; Ullrich, O.; Richter, G.; Thomas, O. Three-point bending behavior of Au nanowires studied by in situ Laue microdiffraction. J. Appl. Phys. 2018, 124, 185104. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.B.; Andriollo, T.; Fæster, S.; Liu, W.; Hattel, J.; Barabash, R.I. Three-dimensional local residual stress and orientation gradients near graphite nodules in ductile cast iron. Acta Mater. 2016, 121, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Barabash, R.I. High resolution mapping of orientation and strain gradients in metals by synchrotron 3D X-ray Laue microdiffraction. Quantum Beam Sci. 2019, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Abboud, A.; Kirchlechner, C.; Send, S.; Micha, J.S.; Ulrich, O.; Pashniak, N.; Strüder, L.; Keckes, J.; Pietsch, U. A new method for polychromatic X-ray μLaue diffraction on a Cu pillar using an energy-dispersive pn-junction charge-coupled device. Rev. Sci. Instrum. 2014, 85, 113901. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, O.; Labat, S.; Cornelius, T.; Richard, M.-I. X-ray Diffraction Imaging of Deformations in Thin Films and Nano-Objects. Nanomaterials 2022, 12, 1363. https://doi.org/10.3390/nano12081363
Thomas O, Labat S, Cornelius T, Richard M-I. X-ray Diffraction Imaging of Deformations in Thin Films and Nano-Objects. Nanomaterials. 2022; 12(8):1363. https://doi.org/10.3390/nano12081363
Chicago/Turabian StyleThomas, Olivier, Stéphane Labat, Thomas Cornelius, and Marie-Ingrid Richard. 2022. "X-ray Diffraction Imaging of Deformations in Thin Films and Nano-Objects" Nanomaterials 12, no. 8: 1363. https://doi.org/10.3390/nano12081363