
Nonmetallic Active Sites on Nickel Phosphide in Oxygen Evolution Reaction




Abstract
:1. Introduction
2. Computational Details
2.1. Static DFT Calculations
2.2. AIMD Simulations
3. Results and Discussion
3.1. Stability of Ni12P5 Surface
3.2. The Adsorption and Dissociation of H2O on the Surface of Ni12P5
- (a)
- Period I: 0–320 fs; H2O approaches the surface of Ni12P5 (001).
- (b)
- Period II: 320–375 fs. H2O is adsorbed and H dissociation starts; the hydrogen bond transforms.
- (c)
- Period III: 375–460 fs; the metastable state, while P-Oa is connected with an H through the hydrogen bond, P-Oa⋯H-Ob.
- (d)
- Period IV: After 460 fs; the breaking of the hydrogen-bond, H+ completely leaves the surface.
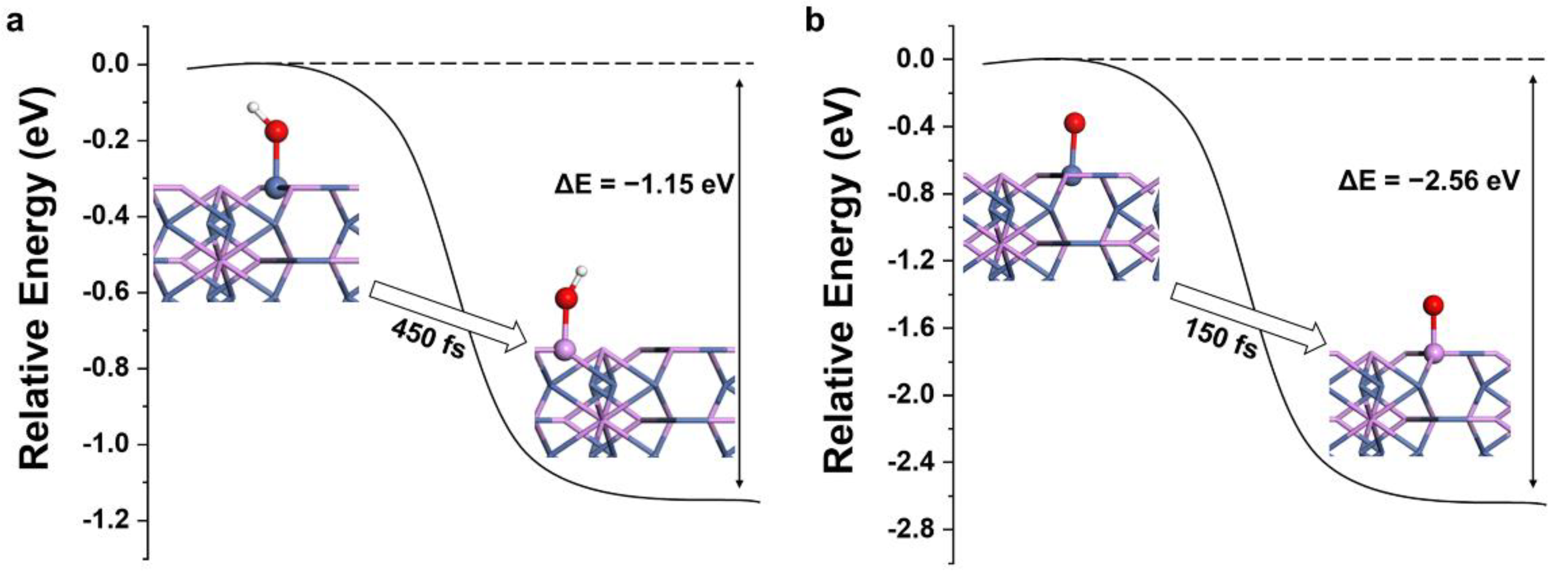
3.3. Nonmetallic P Atoms as Active Sites
3.4. Active Site of OER in Nickel Phosphides
3.5. Charge Distribution at Ni12P5 (001)/H2O
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chow, J.; Kopp, R.J.; Portney, P.R. Energy Resources and Global Development. Science 2003, 302, 1528–1531. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ding, N.; Li, F.; Fan, Y.; Luo, Y.; Li, D.; Meng, Q. Enhancement of photocatalytic H2 evolution on ZnIn2S4 loaded with in-situ photo-deposited MoS2 under visible light irradiation. Appl. Catal. B Environ. 2014, 160–161, 614–620. [Google Scholar] [CrossRef]
- Liu, C.; Röder, R.; Zhang, L.; Ren, Z.; Chen, H.; Zhang, Z.; Ronning, C.; Gao, P.-X. Highly efficient visible-light driven photocatalysts: A case of zinc stannate based nanocrystal assemblies. J. Mater. Chem. A 2014, 2, 4157–4167. [Google Scholar] [CrossRef]
- Wang, C.; Astruc, D. Recent developments of nanocatalyzed liquid-phase hydrogen generation. Chem. Soc. Rev. 2021, 50, 3437–3484. [Google Scholar] [CrossRef]
- Turner, J.A. A Realizable Renewable Energy Future. Science 1999, 285, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; Chang, K.C.; Uchimura, M.; Paulikas, A.P.; Stamenkovic, V.; Markovic, N.M. Enhancing Hydrogen Evolution Activityin Water Splitting by TailoringLi+-Ni(OH)2-Pt Interfaces. Science 2011, 334, 1256–1260. [Google Scholar] [CrossRef]
- Takashima, T.; Hashimoto, K.; Nakamura, R. Inhibition of Charge Disproportionation of MnO2 Electrocatalysts for Efficient Water Oxidation under Neutral Conditions. J. Am. Chem. Soc. 2012, 134, 18153–18156. [Google Scholar] [CrossRef]
- Jiang, N.; You, B.; Sheng, M.; Sun, Y. Electrodeposited Cobalt-Phosphorous-Derived Films as Competent Bifunctional Catalysts for Overall Water Splitting. Angew. Chem. Int. Ed. 2015, 54, 6251–6254. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Q.; Feng, X. Support and Interface Effects in Water-Splitting Electrocatalysts. Adv. Mater. 2019, 31, e1808167. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co 2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Chang, K.C.; Chang, S.H.; Kang, Y.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.T.; Myers, D.; et al. Using surface segregation to design stable Ru-Ir oxides for the oxygen evolution reaction in acidic environments. Angew. Chem. Int. Ed. 2014, 53, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kang, Y.; Huo, Z.; Zhu, Z.; Huang, W.; Xin, H.L.; Snyder, J.D.; Li, D.; Herron, J.A.; Mavrikakis, M.; et al. Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 2014, 343, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef] [Green Version]
- Yamada, I.; Takamatsu, A.; Asai, K.; Ohzuku, H.; Shirakawa, T.; Uchimura, T.; Kawaguchi, S.; Tsukasaki, H.; Mori, S.; Wada, K.; et al. Synergistically Enhanced Oxygen Evolution Reaction Catalysis for Multielement Transition-Metal Oxides. ACS Appl. Energy Mater. 2018, 1, 3711–3721. [Google Scholar] [CrossRef]
- Wang, Y.-N.; Yang, Z.-J.; Yang, D.-H.; Zhao, L.; Shi, X.-R.; Yang, G.; Han, B.-H. FeCoP2 Nanoparticles Embedded in N and P Co-doped Hierarchically Porous Carbon for Efficient Electrocatalytic Water Splitting. ACS Appl. Mater. Interfaces 2021, 13, 8832–8843. [Google Scholar] [CrossRef]
- Liu, M.; Sun, Z.; Li, S.; Nie, X.; Liu, Y.; Wang, E.; Zhao, Z. Hierarchical superhydrophilic/superaerophobic CoMnP/Ni2P nanosheet-based microplate arrays for enhanced overall water splitting. J. Mater. Chem. A 2021, 9, 22129–22139. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, J.; Yang, W.; Lu, B.; Tang, J. Modifying candle soot with FeP nanoparticles into high-performance and cost-effective catalysts for the electrocatalytic hydrogen evolution reaction. Nanoscale 2015, 7, 4400–4405. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, A.; Duan, S.; Zhao, H.; Wang, R.; Lau, W.-M. Au@Co2P core/shell nanoparticles as a nano-electrocatalyst for enhancing the oxygen evolution reaction. RSC Adv. 2019, 9, 40811–40818. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Chen, S.; Ren, J.; Jia, Y.A.; Chen, C.; Komarneni, S.; Yang, D.; Yao, X. Electronic Structure Tuning in Ni3FeN/r-GO Aerogel toward Bifunctional Electrocatalyst for Overall Water Splitting. ACS Nano 2018, 12, 245–253. [Google Scholar] [CrossRef]
- Yang, Y.; Kang, Y.; Zhao, H.; Dai, X.; Cui, M.; Luan, X.; Zhang, X.; Nie, F.; Ren, Z.; Song, W. An Interfacial Electron Transfer on Tetrahedral NiS2/NiSe2 Heterocages with Dual-Phase Synergy for Efficiently Triggering the Oxygen Evolution Reaction. Small 2020, 16, e1905083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, Y.; Zhu, H.; Lu, S.; Ma, P.; Dong, W.; Duan, F.; Chen, M.; Du, M. Understanding the Role of Nanoscale Heterointerfaces in Core/Shell Structures for Water Splitting: Covalent Bonding Interaction Boosts the Activity of Binary Transition-Metal Sulfides. ACS Appl. Mater. Interfaces 2020, 12, 6250–6261. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Duan, S.; Zhu, Y.; Yin, P.; Wang, A.R. Enhanced OER Performances of Au@NiCo2S4 Core-Shell Heterostructure. Nanomaterials 2020, 10, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Lv, Y.; Yin, P.; Zhu, Y.; Wang, R. Constructing the Au–CoNi2S4 core–shell heterostructure to promote the catalytic performance for oxygen evolution. J. Phys. D Appl. Phys. 2021, 54, 425501. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, J.; Sun, Z. Novel 2D Transition-Metal Carbides: Ultrahigh Performance Electrocatalysts for Overall Water Splitting and Oxygen Reduction. Adv. Funct. Mater. 2020, 30, 2000570. [Google Scholar] [CrossRef]
- Stern, L.-A.; Feng, L.; Song, F.; Hu, X. Ni2P as a Janus catalyst for water splitting: The oxygen evolution activity of Ni2P nanoparticles. Energy Environ. Sci. 2015, 8, 2347–2351. [Google Scholar] [CrossRef]
- Lai, C.; Liu, X.; Deng, Y.; Yang, H.; Jiang, H.; Xiao, Z.; Liang, T. Rice-shape nanocrystalline Ni5P4: A promising bifunctional electrocatalyst for hydrogen evolution reaction and oxygen evolution reaction. Inorg. Chem. Commun. 2018, 97, 98–102. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Z.; Cai, C.; Wang, M.; Zhao, Z.L.; Li, M.; Huang, X.; Han, S.; Zhou, H.; Feng, Z.; et al. Single Iridium Atom Doped Ni2P Catalyst for Optimal Oxygen Evolution. J. Am. Chem. Soc. 2021, 143, 13605–13615. [Google Scholar] [CrossRef]
- Riyajuddin, S.; Azmi, K.; Pahuja, M.; Kumar, S.; Maruyama, T.; Bera, C.; Ghosh, K. Super-Hydrophilic Hierarchical Ni-Foam-Graphene-Carbon Nanotubes-Ni2P-CuP2 Nano-Architecture as Efficient Electrocatalyst for Overall Water Splitting. ACS Nano 2021, 15, 5586–5599. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, Y.-R.; Hsu, C.-S.; Xue, H.-G.; Chan, T.-S.; Suen, N.-T.; Chen, H.M. Electronic structure inspired a highly robust electrocatalyst for the oxygen-evolution reaction. Chem. Commun. 2020, 56, 8071–8074. [Google Scholar] [CrossRef]
- Menezes, P.; Indra, A.; Das, C.; Walter, C.; Göbel, C.; Gutkin, V.; Schmeiβer, D.; Driess, M. Uncovering the Nature of Active Species of Nickel Phosphide Catalysts in High-Performance Electrochemical Overall Water Splitting. ACS Catal. 2016, 7, 103–109. [Google Scholar] [CrossRef]
- Xu, Y.; Duan, S.; Li, H.; Yang, M.; Wang, S.; Wang, X.; Wang, R. Au/Ni12P5 core/shell single-crystal nanoparticles as oxygen evolution reaction catalyst. Nano Res. 2017, 10, 3103–3112. [Google Scholar] [CrossRef]
- Sun, H.; Min, Y.; Yang, W.; Lian, Y.; Lin, L.; Feng, K.; Deng, Z.; Chen, M.; Zhong, J.; Xu, L.; et al. Morphological and Electronic Tuning of Ni2P through Iron Doping toward Highly Efficient Water Splitting. ACS Catal. 2019, 9, 8882–8892. [Google Scholar] [CrossRef]
- Zhang, W.-Z.; Chen, G.-Y.; Zhao, J.; Liang, J.-C.; Sun, L.-F.; Liu, G.-F.; Ji, B.-W.; Yan, X.-Y.; Zhang, J.-R. Self-growth Ni2P nanosheet arrays with cationic vacancy defects as a highly efficient bifunctional electrocatalyst for overall water splitting. J. Colloid Interface Sci. 2020, 561, 638–646. [Google Scholar] [CrossRef]
- Yan, H.; Xie, Y.; Wu, A.; Cai, Z.; Wang, L.; Tian, C.; Zhang, X.; Fu, H. Anion-Modulated HER and OER Activities of 3D Ni–V-Based Interstitial Compound Heterojunctions for High-Efficiency and Stable Overall Water Splitting. Adv. Mater. 2019, 31, e1901174. [Google Scholar] [CrossRef]
- Wen, S.; Chen, G.; Chen, W.; Li, M.; Ouyang, B.; Wang, X.; Chen, D.; Gong, T.; Zhang, X.; Huang, J.; et al. Nb-doped layered FeNi phosphide nanosheets for highly efficient overall water splitting under high current densities. J. Mater. Chem. A 2021, 9, 9918–9926. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Pohl, D.; Rellinghaus, B.; Dong, R.; Liu, S.; Zhuang, X.; Feng, X. Interface Engineering of MoS2/Ni3S2 Heterostructures for Highly Enhanced Electrochemical Overall-Water-Splitting Activity. Angew. Chem. Int. Ed. 2016, 55, 6702–6707. [Google Scholar] [CrossRef] [Green Version]
- Skelton, A.A.; Fenter, P.; Kubicki, J.D.; Wesolowski, D.J.; Cummings, P.T. Simulations of the Quartz(10)/Water Interface: A Comparison of Classical Force Fields, Ab Initio Molecular Dynamics, and X-ray Reflectivity Experiments. J. Phys. Chem. C 2011, 115, 2076–2088. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Cuny, J.; Verdolino, V.; Parrinello, M. Aqueous solutions: State of the art in ab initio molecular dynamics. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20120482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-Y.; Le, J.-B.; Wang, Y.-H.; Chen, S.; Yang, Z.-L.; Li, J.-F.; Cheng, J.; Tian, Z.-Q. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 2019, 18, 697–701. [Google Scholar] [CrossRef]
- Réocreux, R.; Jiang, T.; Iannuzzi, M.; Michel, C.; Sautet, P. Structuration and Dynamics of Interfacial Liquid Water at Hydrated γ-Alumina Determined by ab Initio Molecular Simulations: Implications for Nanoparticle Stability. ACS Appl. Nano Mater. 2017, 1, 191–199. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [Green Version]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure. Phys. Rev. B 2001, 65, 035406. [Google Scholar] [CrossRef] [Green Version]
- Wexler, R.B.; Martirez, J.M.P.; Rappe, A.M. Stable Phosphorus-Enriched (0001) Surfaces of Nickel Phosphides. Chem. Mater. 2016, 28, 5365–5372. [Google Scholar] [CrossRef]
- Loh, Z.-H.; Doumy, G.; Arnold, C.; Kjellsson, L.; Southworth, S.H.; Al Haddad, A.; Kumagai, Y.; Tu, M.-F.; Ho, P.J.; March, A.M.; et al. Observation of the fastest chemical processes in the radiolysis of water. Science 2020, 367, 179–182. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; MacKay, L.; Lamoureux, G. Development of Semiempirical Models for Proton Transfer Reactions in Water. J. Chem. Theory Comput. 2014, 10, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Pezeshki, S.; Lin, H. Adaptive-Partitioning QM/MM for Molecular Dynamics Simulations: 4. Proton Hopping in Bulk Water. J. Chem. Theory Comput. 2015, 11, 2398–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhu, J.; Wang, P.; Chen, D.; Zhang, C.; Xiao, M.; Ma, Q.; Bai, H.; Qin, R.; Ma, J.; et al. Fe–Co–P multi-heterostructure arrays for efficient electrocatalytic water splitting. J. Mater. Chem. A 2021, 9, 24677–24685. [Google Scholar] [CrossRef]
- Légaré, M.-A.; Bélanger-Chabot, G.; Dewhurst, R.D.; Welz, E.; Krummenacher, I.; Engels, B.; Braunschweig, H. Nitrogen fixation and reduction at boron. Science 2018, 359, 896–900. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Li, H.; Xiao, J.; Tu, Y.; Deng, D.; Yang, H.; Tian, H.; Li, J.; Ren, P.; Bao, X. Triggering the electrocatalytic hydrogen evolution activity of the inert two-dimensional MoS2 surface via single-atom metal doping. Energy Environ. Sci. 2015, 8, 1594–1601. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Feng, Y.; Allen, C.S.; Wan, C.; Volosskiy, B.; Li, M.; Zhao, Z.; Wang, Y.; Sun, H.; et al. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 2018, 1, 63–72. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, Q.; Luo, Q.; Sheng, L.; Yang, J. Understanding Single-Atom Catalysis in View of Theory. JACS Au 2021, 1, 2130–2145. [Google Scholar] [CrossRef]
- Dong, C.; Li, Y.; Cheng, D.; Zhang, M.; Liu, J.; Wang, Y.-G.; Xiao, D.; Ma, D. Supported Metal Clusters: Fabrication and Application in Heterogeneous Catalysis. ACS Catal. 2020, 10, 11011–11045. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Yin, F.; Chu, W.; Chen, B. Coordinately unsaturated metal–organic framework as an unpyrolyzed bifunctional electrocatalyst for oxygen reduction and evolution reactions. J. Mater. Chem. A 2020, 8, 22111–22123. [Google Scholar] [CrossRef]
- Shah, K.; Dai, R.; Mateen, M.; Hassan, Z.; Zhuang, Z.; Liu, C.; Israr, M.; Cheong, W.-C.; Hu, B.; Tu, R.; et al. Cobalt Single Atom Incorporated in Ruthenium Oxide Sphere: A Robust Bifunctional Electrocatalyst for HER and OER. Angew. Chem. Int. Ed. 2022, 61, e202114951. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, J.; Xie, L.; Wu, J.; Ren, Q.; Wang, Y.; Li, G. Surface-Adsorbed Carboxylate Ligands on Layered Double Hydroxides/Metal–Organic Frameworks Promote the Electrocatalytic Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2021, 60, 18129–18137. [Google Scholar] [CrossRef]
- Wang, T.; Chen, H.; Yang, Z.; Liang, J.; Dai, S. High-Entropy Perovskite Fluorides: A New Platform for Oxygen Evolution Catalysis. J. Am. Chem. Soc. 2020, 142, 4550–4554. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Yang, J.; Liu, M.; Wang, R.; Zhang, G.; Wang, H.; Tang, H.; Liu, C.; Mei, Z.; Chen, H.; et al. Tuning Electronic Push/Pull of Ni-Based Hydroxides To Enhance Hydrogen and Oxygen Evolution Reactions for Water Splitting. ACS Catal. 2018, 8, 5621–5629. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhou, J.; Han, L.; Sun, C.; Wang, Q.; Su, Z. Ternary hybrids as efficient bifunctional electrocatalysts derived from bimetallic metal–organic-frameworks for overall water splitting. J. Mater. Chem. A 2018, 6, 5789–5796. [Google Scholar] [CrossRef]
- Chu, S.; Sun, H.; Chen, G.; Chen, Y.; Zhou, W.; Shao, Z. Rationally designed Water-Insertable Layered Oxides with Synergistic Effect of Transition-Metal Elements for High-Performance Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2019, 11, 25227–25235. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Charge/e− | Eads/eV | Charge/e− | Eads/eV | ||
|---|---|---|---|---|---|
| *OH | 0.37 | 2.54 | *O | 0.15 | 1.24 |
| 0.85 | 1.71 | 0.21 | 0.92 | ||
| 0.89 | 1.42 | 0.21 | 0.82 | ||
| 0.91 | 1.31 | 0.40 | 0.69 | ||
| 0.94 | 0.51 | 0.43 | 0.50 | ||
| 0.95 | 0.53 | 0.44 | 0.45 |
| Structures | Formulas |
|---|---|
| Ni*OH | |
| Ni*O | |
| P*OH | |
| P*O |
| H2O | Ni | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| P | O | H1 | H2 | Ni1 | Ni2 | Ni3 | Ni4 | Ni5 | |
| *+H2O | −0.18 | −1.11 | 0.55 | 0.56 | 0.21 | 0.21 | 0.11 | 0.12 | 0.08 |
| *OH2 | 0.66 | −1.56 | 0.65 | 0.60 | 0.17 | 0.11 | 0.12 | 0.10 | 0.08 |
| *OH + H | 0.87 | −1.67 | 0.61 | 0.59 | 0.07 | 0.29 | 0.06 | 0.06 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Qiu, H.; Li, H.; He, J.; Xu, Y.; Wang, R. Nonmetallic Active Sites on Nickel Phosphide in Oxygen Evolution Reaction. Nanomaterials 2022, 12, 1130. https://doi.org/10.3390/nano12071130
Zhang P, Qiu H, Li H, He J, Xu Y, Wang R. Nonmetallic Active Sites on Nickel Phosphide in Oxygen Evolution Reaction. Nanomaterials. 2022; 12(7):1130. https://doi.org/10.3390/nano12071130
Chicago/Turabian StyleZhang, Pengfei, Hongmei Qiu, Huicong Li, Jiangang He, Yingying Xu, and Rongming Wang. 2022. "Nonmetallic Active Sites on Nickel Phosphide in Oxygen Evolution Reaction" Nanomaterials 12, no. 7: 1130. https://doi.org/10.3390/nano12071130